June Newsletter: Kinesin Motor Proteins and Neurodegeneration
Introduction
Kinesin motor proteins regulate mitosis and anterograde cargo transport as exemplified by fast axonal transport (FAT) in neurons. Neurons depend on kinesins for cell cycle regulation, especially the assembly and function of the mitotic spindle, a macromolecular structure composed primarily of microtubules (MTs) that undergo cycles of polymerization and depolymerization to properly segregate duplicate chromosomes into separate daughter cells. In addition, the kinesin motors use MTs to transport cargo such as proteins, lipids, and mitochondria to the axon and axon terminal, an essential process for normal neuron development, growth, and communication as the cell body is the site of synthesis1.
In neurodegenerative diseases such as Alzheimer’s disease (AD), cell cycle defects (e.g., chromosome mis-segregation, abnormal mitotic spindle structure/function, aneuploidy) and impaired FAT are pathophysiological hallmarks of AD, the most common form of dementia2,3. Indeed, FAT deficits and axonal swellings occur before the classic neuropathological signs of dementia (plaques and tangles), leading some researchers to posit that transport deficits are an early sign of neuron vulnerability to neurodegeneration4-8. This newsletter focuses on the role Eg5 (KSP, kinesin spindle protein, KIF11, kinesin-5) and kinesin-1 have in AD-associated cell cycle and FAT defects, respectively. Eg5 is a kinesin motor that interacts with MTs and is essential for mitotic spindle formation and function. Kinesin-1 is the prototypical kinesin that mediates FAT. For a discussion of the role of the dynein motor in neurodegeneration, see the 2014 January/February newsletter.

Motors and Neurodegeneration: Cell Cycle Defects
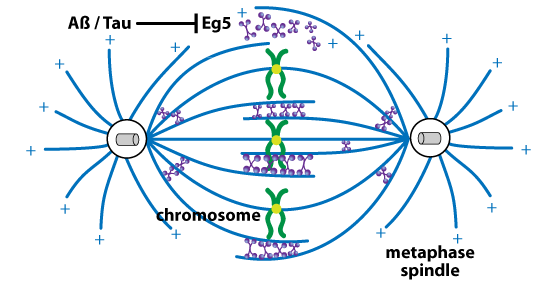
The neuropathological hallmarks of AD are beta amyloid (Aß) plaques and hyperphosphorylated tau neurofibrillary tangles. Aß is produced by enzymatic cleavage of amyloid precursor protein (APP) by beta-secretase 1 (BACE1) and the presenilin (PS)-containing gamma-secretase complex2,3. In AD-associated cell cycle defects, both Aß and tau inhibit Eg5 activity and its interaction with MTs, which negatively affects mitotic spindle structure and function9-11 (Fig. 1). A dysfunctional spindle results in the mis-segregation of chromosomes, aneuploidy/hyperploidy, and cell death9-11. Tau's inhibition of Eg5 requires excess levels of MT-bound tau10. Neurons displaying aneuploidy/hyperploidy are increased in preclinical AD and selectively die as AD progresses. Indeed, 90% of cell death in autopsied AD brains is comprised of hyperploid neurons12.
Eg5’s role in AD is not limited to cell cycle defects as mature, post-mitotic neurons express this protein. Eg5 mediates Aß-induced inhibition of long-term potentiation and loss of hippocampal spines/synapses13,14, at least partly through a reduction in cell surface trafficking of NMDA and nerve growth factor/p75 neutrophin receptors13.
Motors and Neurodegeneration: Axonal Transport Defects
All 4 neurodegeneration-related proteins (APP, Aß, PS, and tau) inhibit kinesin-1-mediated FAT. For APP and Aß disruption of FAT15-17, various mechanisms have been reported, ranging from actin aggregation and dynamic changes in actin polymerization16 to activation of casein kinase 2, which initiates a sequential cascade of kinesin-1 light chain phosphorylation and release of cargo from kinesin-117. Interestingly, APP undergoes anterograde FAT18 via binding with kinesin-1 light chains as part of an axon membrane compartment which also contains BACE1 and PS119-21. In this way, APP could be cleaved into Aß during and/or after kinesin-1-mediated FAT21. However, this finding has been refuted by others22.
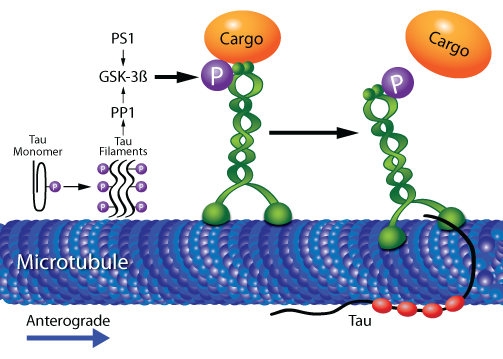
Both tau and mutant PS1 (M146V) regulate kinesin-1-mediated FAT via dephosphorylation-stimulated activation of GSK-3ß, resulting in GSK-3ß-mediated phosphorylation of kinesin-1 light chains, which in turn induce separation of kinesin-1 and its cargo5 (Fig. 2). As a tau kinase, GSK-3ß can mediate formation of pathological, hyper-phosphorylated tau, which dissociates from MTs and causes MT depolymerization, followed by formation of tau filaments and eventually neurofibrillary tangles2,3. The amino terminus of either tau filaments or C-terminal-truncated, unbound monomers (C-terminus contains MT binding domains) inhibits kinesin-dependent FAT. Here, GSK-3ß activity is increased via protein phosphatase 1 (PP1)-mediated dephosphorylation-induced activation of GSK-3ß23 (Fig. 2). These studies have led researchers to suggest that FAT is vulnerable to disruption early in the pathophysiology of AD2-5; indeed, axonal/transport defects occur long before amyloid deposition4. Furthermore, depletion of kinesin-1 produces similar transport deficits while also increasing levels of Aß peptide levels and deposits4.
Conclusion
Normal neuron physiology relies on kinesin motors for a variety of processes, including proper mitosis and FAT. Impaired kinesin function has a profound and wide-spread effect on neuron health as kinesin motor dysfunctions are not only observed in AD, but Huntington's disease24, upper and lower motor neuron diseases, and Charcot-Marie-Tooth peripheral neuropathy2,3. To assist in studying how kinesin and dynein motor proteins regulate neuron health and survival, Cytoskeleton, Inc. offers purified kinesin motor proteins and dynein protein, along with kits to measure MT-induced motor ATPase activity.
Related Products & Resources
Kinesin and Dynein Proteins
CENP-E kinesin motor domain protein: GST tagged: Homo sapiens recombinant (Cat. # CP01)
CHromokinesin kinesin motor domain protein: GST tagged: Homo sapiens recombinant (Cat. # CR01)
Cytoplasmic Dynein Motor Protein (Cat. # CS-DN01)
Eg5 kinesin motor domain protein: GST tagged: Homo sapiens recombinant (Cat. # EG01)
KIFC3 kinesin motor domain protein: GST tagged: Homo sapiens recombinant (Cat. # KC01)
KIF3C kinesin motor domain protein: GST tagged: Homo sapiens recombinant (Cat. # KF01)
KIF7 Motor Domain Protein: Homo sapien (Cat. # CS-KF51)
Kinesin heavy chain motor domain protein: GST tagged: Homo sapiens recombinant (Cat. # KR01)
MCAK kinesin motor domain protein: GST tagged: Homo sapiens recombinant (Cat. # MK01)
MKLP kinesin motor domain protein: GST tagged: Homo sapiens recombinant (Cat. # MP01)
MKLP2 Motor Domain Protein (Cat. # CS-MP05)
Kits and Assays
ATPase/GTPase Kinetic ELIPA Assay Kit (Cat. # BK051/052)
Cytophos Endpoint Phosphate Assay (Cat. # BK054)
Kinesin ELIPA Kit (Cat. # BK060)
HTS kinesin ATPase Endpoint Assay Biochem Kit (Cat. # BK053)
Microtubules
Tubulin protein (pre-formed microtubules): porcine brain (Cat. # MT002)
References
- Bass P.W. 1998. The role of motor proteins in establishing the microtubule arrays of axons and dendrites. J. Chem. Neuroanat. 14, 175-180.
- Millecamps S. and Julien J.-P. 2013. Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 14, 161-176.
- Morfini G.A. et al. 2009. Minisymposium: Axonal transport defects in neurodegenerative diseases. J. Neurosci. 29, 12776-12786.
- Stokin G.B. et al. 2005. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science. 307, 1282-1288.
- Pigino G. et al. 2003. Alzheimer’s presenilin 1 mutations impair kinesin-based axonal transport. J. Neurosci. 23, 4499-4508.
- Lazarov O. et al. 2007. Impairments in fast axonal transport and motor neuron deficits in transgenic mice expressing familial Alzheimer’s disease-linked mutant presenilin 1. J. Neurosci. 27, 7011-7020.
- Ishihara T. et al. 1999. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron. 24, 751-762.
- Zhang B. et al. 2004. Retarded axonal transport of R406W mutant tau in transgenic mice with a neurodegenerative tauopathy. J. Neurosci. 24, 4657-4667.
- Borysov S.I. et al. 2011. Alzheimer Aß disrupts the mitotic spindle and directly inhibits mitotic microtubule motors. Cell Cycle. 10, 1397-1410.
- Bouge A.-L. and Parmentier M.-L. 2016. Tau excess impairs mitosis and kinesin-5 function, leading to aneuploidy and cell death. Dis. Model. Mech. 9, 307-319.
- Rossi G. et al. 2008. A new function of microtubule-associated protein tau: involvement in chromosome stability. Cell Cycle. 7, 1788-1794.
- Arendt T. et al. 2010. Selective cell death of hyperploid neurons in Alzheimer’s disease. Am J. Pathol. 177, 15-20.
- Ari C. et al. 2014. Alzheimer Aß inhibition of eg5/kin5 reduces neurotrophin/transmitter receptor function. Neurobiol. Aging. 35, 1839-1849.
- Freund R.K. et al. 2016. Inhibition of the motor protein Eg5/Kinesin-5 in amyloid b-mediated impairment of hippocampal long-term potentiation and dendritic spine loss. Mol. Pharmacol. 89, 552-559.
- Gunawardena S. and Goldstein L.S.B. 2001. Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron. 32, 389-401.
- Hiruma H. et al. Glutamate and amyloid b-protein rapidly inhibit fast axonal transport in cultured rat hippocampal neurons by different mechanisms. J. Neurosci. 23, 8967-8977.
- Pigino G. et al. 2009. Disruption of fast axonal transport is a pathogenic mechanism for intraneuronal amyloid beta. Proc. Natl. Acad. Sci. USA. 106, 5907-5912.
- Koo E.H. et al. 1990. Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc. Natl. Acad. Sci. USA. 87, 1561-1565.
- Ferreira A. et al. 1993. Intraneuronal compartments if the amyloid precursor protein. J. Neurosci. 13, 3112-3123.
- Kamal A. et al. 2000. Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron. 28, 449-459.
- Kamal A. et al. 2001. Kinesin-mediated axonal transport of a membrane compartment containing ß-secretase and presenilin-1 requires APP. Nature. 414, 643-648.
- Lazarov O. et al. 2005. Axonal transport, amyloid precursor protein, kinesin-1, and the processing apparatus: Revisited. J. Neurosci. 25, 2386-2395.
- LaPointe N.E. et al. 2009. The amino terminus of tau inhibits kinesin-dependent axonal transport: Implications for filament toxicity. J. Neurosci. Res. 87, 440-451.
- Morfini G.A. et al. 2009. Pathogenic Huntingtin inhibits fast axonal transport by activating JNK3 and phosphorylating kinesin. Nat. Neurosci. 12, 864-871.