Actin Modifications and the Cytoskeleton
Actin, a highly expressed and ubiquitous cytoskeletal protein, is a major substrate for at least 17 post-translational modifications (PTMs)1. PTMs are highly dynamic and often reversible processes where a protein’s functional properties are altered by addition of a chemical group or another protein to its amino acid residues. With roles in cell growth, motility, trafficking, and division, it is imperative to understand how actin’s function is altered by PTMs. The aim of this newsletter is to summarize what is known about 3 important actin PTMs: arginylation, glutathionylation, and phosphorylation (Fig. 1).
Arginylation
Arginylation is mediated by arginyltransferase (Ate1) and involves the addition of arginine on the N-terminus of beta-actin by a peptide bond2,3. Arginylation can impact actin’s function in several ways. For example, arginylation increases actin polymerization3,4 and strengthens the actin filament network3, the main structural support for maintaining dendritic spine morphology and size5. Blockade/loss of arginylation is associated with defects in cell migration and myofibril contraction4,6-8 as well as collapse of leading edge lamella and reduced F-actin levels3. The leading edge collapse is specifically due to decreased N-terminally arginylated beta actin4. Besides the N-terminus, arginylation also occurs internally on the actin molecule on at least two residues1. Internal arginylation is predicted to affect polymerization and interactions between actin and actin binding proteins1. Ex vivo studies using fibroblasts cultured from Ate1 knockout mice also support a role for arginylation in actin function.

Cells have slower rates of polymerization (faster nucleation/slower elongation), decreased F-actin staining, shorter actin filaments, and an increased number of intracellular actin aggregates1,3 (Fig. 1). In vivo, Ate1 knockout mice have defects in cardiovascular development9 and neural crest morphogenesis7.
Glutathionylation
Glutathionylation is one of many reduction-oxidation (redox) PTMs that target two of actin’s cysteine amino acid residues (Cys217, Cys374)1. Glutathionylation is a reversible PTM whereby glutathione is attached to an actin’s cysteine residue via a disulfide bond, creating glutathione disulfide. Actin glutathionylation serves to protect actin, and thereby cells, from oxidative stress10-12. For example, actin glutathionylation is believed to participate in stabilization of axons and dendrites as well as neuron survival during periods of oxidative stress11. Furthermore, actin glutathionylation influences how cells’ actin networks respond to growth factors, mediating actin polymerization and subsequent trafficking and re-arrangement of F-actin13. During oxidative stress, glutathionylation increases which decreases actin polymerization, resulting in reduced F-actin levels1,13 (Fig. 1). Besides inhibiting F-actin formation, increased glutathionylation has also been linked to abnormal rearrangement of actin filaments12,14. Upon reversal of glutathionylation, actin polymerization increases13.
Phosphorylation
Actin has at least 35 amino acid residues that can be modified by phosphorylation and this PTM can exert both negative and positive effects on polymerization1. For example, when actin’s Tyr53 residue in the slime mold Dictyostelium is phosphorylated, polymerization decreases, likely through a disruption of actin subunit-subunit contact15,16. Conversely, in the slime mold Physarum, actin fragmin kinase (AFK), a calcium-dependent enzyme, phosphorylates actin’s Thr201-203 residues, leading to elongation of the actin filaments17-20. This elongation is believed to be a result of reduced interactions between fragmin and actin. Fragmin is related to the severing protein gelsolin and as such, controls filament length17-20. The effect of Thr phosphorylation is reversed by protein phosphatases PP1 and PP2A21. In both organisms, the changes in actin phosphorylation states are associated with cytoskeletal responses to extracellular events (e.g., locomotion, phagocytosis, signal transduction) and transition into a state of dormancy1,15-18.
In mammals, proteomic analyses have revealed that multiple kinases phosphorylate actin and vary by cell type, disease conditions, and external stimuli. Unfortunately, many of the studies are correlational and do not report a direct relationship between a given kinase and actin phosphorylation1. For example, Ser and Tyr residues on actin are phosphorylated in response to insulin via unknown kinases, leading to reduced DNAse I binding1 (Fig. 1). Likewise, activation of the p21-activated kinase PAK1 leads to actin phosphorylation which is correlated with loss of stress fibers and altered F-actin localization22. Similarly, Src kinase-driven phosphorylation of actin impairs actin polymerization1,23. Several known actin kinases are casein kinase I1,24, cAMP-dependent protein kinase (PKA), and calcium/phosphoinositide-dependent protein kinase (PKC)25,26. Casein kinase I phosphorylates actin similar to AFK (targets Thr and Ser residues and is calcium-dependent). PKA and PKC act in an opposing manner with the former impairing polymerization and the latter stimulating it27,28 (Fig. 1).
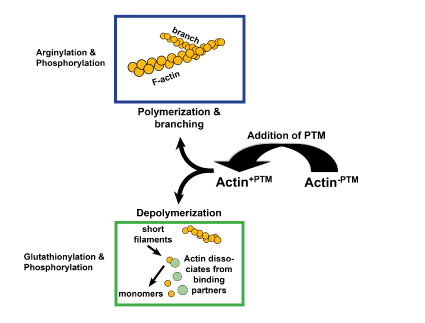
Figure 1: Post-translational modifications (PTMs) affect actin activity differentially. Arginylation promotes polymerization while glutathionylation decreases it. Phosphorylation can either increase or decrease actin polymerization, depending on the residue modified. Phosphorylation also affects actin binding.
In summary, actin is a major cytoskeletal protein whose function is modulated by a variety of PTMs. Despite actin’s relevance in all aspects of cell biology, our current understanding of how at least 17 different PTMs affect actin polymerization, stability, and binding is not complete. As new PTM tools are developed, we can look forward to greatly advancing our understanding of PTMs not only for actin, but for many other cytoskeletal proteins.
Actin Related Research Tools
| Protein | Source | Purity | Cat. # | Amount | ||||
Actin Protein | Rabbit skeletal muscle | >99% | 4 x 250 µg | |||||
Actin Protein | Porcine Brain | >99% | 2 x 250 µg | |||||
Pre-formed Actin Filaments | Rabbit skeletal muscle | >99% | 1 x 1 mg | |||||
| Pyrene Actin Protein | Rabbit skeletal muscle | >99% | AP05-A AP05-B | 1 x 1 mg 5 x 1 mg | ||||
| Biotinylated Actin Protein | Rabbit skeletal muscle | >99% | 5 x 20 mg | |||||
| Kit | Cat. # | Amount | ||||||
G-actin/F-actin In Vivo Biochem Kit™ | BK037 | 30-100 assays | ||||||
Actin Binding Protein Spin-down Assay Biochem Kit™ | BK013 | 30-100 assays | ||||||
Actin Polymerization Biochem Kit™ | BK003 | 24-30 assays | ||||||
References
- J.R. Terman and A. Kashina, 2013. Post-translational modification and regulation of actin. Curr. Opin. Cell Biol. 25, 1-9.E.
- Balzi et al., 1990. Cloning and functional analysis of the arginyl-tRNA-protein transferase gene ATE1 of Saccharomyces cerevisiae. J. Biol. Chem. 265, 7464-7471.
- S. Saha et al., 2010. Arginylation regulates intracellular actin polymer level by modulating actin properties and binding of capping and severing proteins. Mol. Biol. Cell. 21, 1350-1361.
- M. Karakozova et al., 2006. Arginylation of beta-actin regulates actin cytoskeleton and cell motility. Science. 313, 192-196.
- P. Hotulainen et al., 2009. Defining mechanisms of actin polymerization and depolymerization during dendritic spine morphogenesis. J. Cell Biol. 185, 323-339.
- R. Rai et al., 2008. Arginyltransferase regulates alpha cardiac actin function, myofibril formation and contractility during heart development. Development. 135, 3881-3889.
- S. Kurosaka et al., 2010. Arginylation-dependent neural crest cell migration is essential for mouse development. PLoS Genet. 6, e10000878.
- S. Kurosaka et al., 2012. Arginylation regulates myofibrils to maintain heart function and prevent dilated cardiomyopathy. J. Mol. Cell Cardiol. 53, 333-341.
- Y.T. Kwon et al., 2002. An essential role of N-terminal arginylation in cardiovascular development. Science. 297, 96-99.
- I. Dalle-Donne et al., 2005. S-glutathionylation in human platelets by a thiol-disulfide exchange-independent mechanism. Free Radic. Biol. Med. 38, 1501-1510.
- M. Sparaco et al., 2006. Protein glutathionylation in human central nervous system: potential role in redox regulation of neuronal defense against free radicals. J. Neurosci. Res. 83, 256-263.
- E.A. Sabens Liedhegner et al., 2012. Mechanisms of altered redox regulation in neurodegenerative diseases—Focus on S-glutathionylation. Antioxid. Redox Signal. 16, 543-566.
- J. Wang et al., 2003. Stable and controllable RNA interference: Investigating the physiological function of glutathionylated actin. Proc. Natl. Acad. Sci. USA. 100, 5103-5106.
- A. Pastore et al., 2003. Actin glutathionylation increases in fibroblasts of patients with Friedreich’s ataxia. J. Biol. Chem. 278, 2588–42595.
- X. Liu et al., 2006. Phosphorylation of actin Tyr-53 inhibits filament nucleation and elongation and destabilizes filaments. Proc. Natl. Acad. Sci. USA. 103, 13694-13699.
- K. Baek et al., 2008. Modulation of actin structure and function by phosphorylation of Tyr-53 and profilin binding. Proc. Natl. Acad. Sci. USA. 105, 11748-11753.
- B. Constantin et al., 1998. Disruption of the actin cytoskeleton of mammalian cells by the capping complex actin-fragmin is inhibited by actin phosphorylation and regulated by Ca2+ ions. J. Cell Sci. 111, 1695-1706.
- K. Furuhasi and S. Hatano, 1990. Control of actin filament length by phosphorylation of fragmin-actin complex. J. Cell Biol. 111, 1081-1087.
- K. Furuhasi and S. Hatano, 1992. Identification of actin kinase activity in purified fragmin-actin complex. FEBS Lett. 310, 34-36.
- S. Steinbacher et al., 1999. The crystal structure of the Physarum polycephalum actin-fragmin kinase: an atypical protein kinase with a specialized substrate-binding domain. EMBO J. 18, 2923-2929.
- E. Waelkens et al., 1995. Microfilament dynamics: regulation of actin polymerization by actin-fragmin kinase and phosphatases. Adv. Enzyme Regul. 35, 199-227.
- E.A. Papakonstanti and C. Stournaras, 2002. Association of PI-3 kinase with PAK1 leads to actin phosphorylation and cytoskeletal reorganization. Mol. Biol. Cell. 13, 2946-2962.
- C.A. Hirshman et al., 2005. Isoproterenol induces actin depolymerization in human airway smooth muscle cells via activation of an Src kinase and Gs. Am. J. Physiol. Lung Cell Mol. Physiol. 288, L924-L931.
- T. Shibayama et al., 1986. Phosphorylation of muscle and non-muscle actins by casein kinase 1 in vitro. Biochem. Int. 13, 367-373.
- J.M. Carrascosa and O.H. Wieland, 1986. Evidence that (a) serine specific protein kinase(s) different from protein kinase C is responsible for the insulin-stimulated actin phosphorylation by placental membrane. FEBS Lett. 201, 81-86.
- M.P. Walsh et al., 1981. Phosphorylation of smooth muscle actin by the catalytic subunit of the cAMP-dependent protein kinase. Biochem. Biophys. Res. Comm. 102, 149-157
- A.G. Prat et al., 1993. Activation of epithelial Na+ channels by protein kinase A requires actin filaments. Am. J. Physiol. 265, C224-C233.
- Y. Ohta et al., 1987. Protein kinase C and cAMP-dependent protein kinase induce opposite effects on actin polymerizability. FEBS Lett. 222, 305-310.