
Cytoskeleton, Inc. was founded in 1993 and coincidentally that was the era when the Rho family of small G-proteins was determined to be major regulators of cytoskeletal dynamics. This finding was established in a very direct way by the seminal paper authored by Drs. Anne Ridley and Alan Hall who at that time worked at the Medical Research Council in London(1). Since then, the field has grown exponentially and now the annual publication rate in this area is approximately 750 papers per year(2). In the intervening years, Alan was always available for consultation; for example, he wrote a mini-review for Cytoskeleton’s 1999 catalog(3). This giving character embodied his spirit and was emphasized in the various obituaries written by many colleagues. Alan’s legacy will continue through his influential body of work, and through the many students and colleagues whom he mentored.
Dr. Hall’s laboratory published two papers in the early part of 2015, both focused on the mechanisms involved in the creation and maintenance of intercellular connections in bronchial epithelial cells. The first paper described a new cross-talk mechanism between the Ras GEF SOS1, PAK4, a member of the serine/threonine p21-activating kinase family, and the scaffold/polarity protein Par6B4. Both PAK4 and Par6B are downstream of Cdc42, a Rho family GTPase.

GTP-bound, active Cdc42 can bind to PAK4 , which would presumably activate this kinase. Here, Hall and colleagues(4) demonstrate that PAK4 phosphorylates Par6B, with the primary site being the Ser143 residue. Interestingly, the Hall laboratory found that constitutively active Cdc42 (ca-Cdc42; Cdc42V12) binds to wild-type and a non-phosphorylatable Par6B mutant (S143A) in cell lysates, but ca-Cdc42 does not bind to a phosphomimetic Par6B mutant (S143E). This provokes the question as to whether Cdc42, being a prenylated cell polarity protein, is in a position to bind and localize the unphosphorylated scaffold protein at the plasma membrane (plm), and function to assist tight junction assembly (see Figure 2). Likewise, PAK4 could be localized to the plm via Cdc42, placing it in the correct location to phosphorylate Par6B. In this situation, Cdc42 functions to maintain tight junctions by causing Par6B phosphorylation, which presumably can no longer be re-localized because its Cdc42-binding capability has been disabled. Thus, Cdc42 could be regulating the assembly and maintenance of tight junctions by coordinating the activity and function of these two downstream targets.
Figure 2: Molecular mechanisms of tight junction assembly.
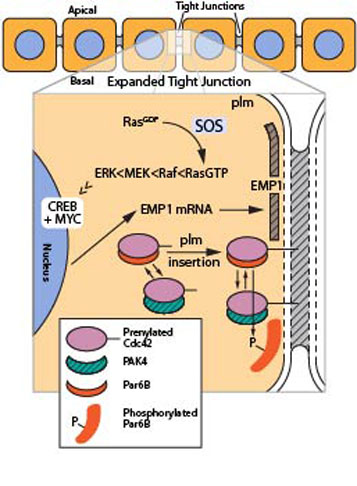
Legend: Schematic image depicting two pathways that help assemble a tight junction complex. Notice the cross-talk mechanism of the Cdc42/PAK4/Par6B interactions compared to the linear pathway of SOS1/Ras/MEK/ERK.
The second paper, citing a more traditional linear pathway from SOS1 > Ras > MEK > ERK, describes a different, but complimentary, mechanism to support tight junction assembly(5). The paper describes setting up a cell-based screen of 87 GEF proteins whose expression has been reduced by RNA interference (RNAi) followed by analysis of each sample’s phenoptype using immunofluorescence with the ZO-1 antibody, a tight junction specific marker. Cells that lost the ability to assemble tight junctions showed disrupted peripheral staining and had an irregular shape. siRNA-mediated knockdown of the SOS1 GEF was one of two GEFs that resulted in disrupted staining; the other GEF already known to be involved is p114RhoGEF. The SOS1 pathway was investigated by using the same phenotypic assay with cells in the presence of either a) a transfected dominant negative Ras gene, b) a MEK inhibitor (GSK1120212), or c) a ERK inhibitor (SCH772984). In all cases, the phenotype was highly disrupted, but strangely the MEK inhibitor only showed strong effects after chronic exposure (4 day treatment) rather than a short 4 hour treatment which did not inhibit junction assembly. Thus, Dr. Hall and colleagues hypothesized that gene expression might be involved, so they screened all the disruptive conditions via microarray with 47,000 genetic markers. 33 genes showed significant reduction in expression, including known Ras>Raf>MEK>ERK>CREB/MYC-regulated genes and several genes connected with tight junctions, including epithelial membane protein 1 (EMP1), a scaffold protein.
In conclusion, Dr. Hall’s laboratory was developing genetic and molecular approaches to investigate how epithelial cell barriers are established by assembling tight junctions via multiple processes involving at least two signal transduction pathways. Dr. Alan Hall will surely be missed in this field, but hopefully his research will continue through the work of others.
References
1. Ridley A. and Hall A. 1992. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 70, 389-399.
2. Pubmed citation search for 2014 using “rho gtpase” as search terms.
3. Hall A. 1999. Rho GTPases and the actin cytoskeleton. Cytoskeleton, Inc. Catalog, 1999, p5-7.
4. Jin D. et al. 2015. Functional cross-talk between Cdc42 and two downstream targets, Par6B and PAK4. Biochem. J. 467, 293-302.
5. Durgan J. et al. 2015. SOS1 and Ras regulate epithelial tight junction formation in the human airway through EMP1. EMBO Rep. 16, 87-96.