Dynein: One Motor, Multiple Neurodegenerative Diseases
Cytoplasmic dynein 1 (hereafter referred to as dynein) is a 1.6 MDa multi-protein complex that serves as the primary ATP-hydrolyzing motor responsible for retrograde axonal transport along microtubules (MTs) in eukaryotic cells (Fig. 1A). Additionally, dynein is essential for many other cellular processes, including mitochondrial movement, endosomal and lysosomal trafficking, transporting mis-folded proteins bound for degradation, nuclear positioning, and mitosis 1-3 (Fig. 2).
The dynein multi-protein complex consists of two ~530 kDa heavy chains that homodimerize along with multiple intermediate, light-intermediate, and light chains (Fig. 1A). The homodimer heavy chain heads are ATPases which provide the energy for movement along the MTs . Binding of the dynein complex to cargo and dynein’s processivity is dependent upon dynein’s omni-present binding protein dynactin1-3 (Fig. 1B) and specific adaptor proteins such as presenilin, LIS1, NUDEL, MuMA, Miro, Milton, BimL, and BimEL1-3.
Mutations in dynein (or dynactin) underlie some neurodegenerative diseases in humans, manifested by axonal transport defects, neuron degeneration, locomotor abnormalities, and/or other neural deficits4-8 (Figs. 1A, 1B). Likewise, animals modeling dynein dysfunction or mutations display motor and neural impairments similar to these human patients3,9-11 (Fig. 1A). The observed neuron loss, protein aggregation, and/or axonal transport deficits are all pathophysiological hallmarks of neurodegeneration, thus motivating further examination of dynein’s role(s) in various neurodegenerative diseases (Fig. 2).

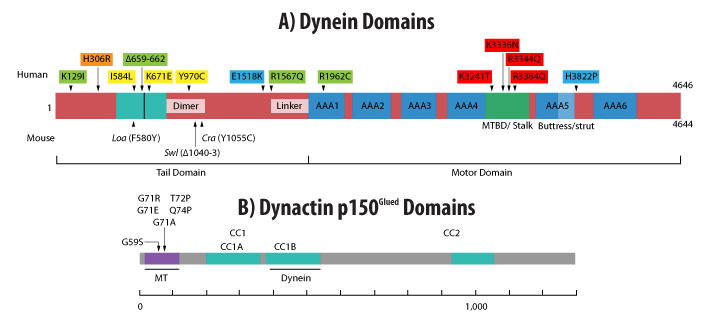
Figure 1: Domains of cytoplasmic dynein heavy chain (A) and dynactin p150Glued (B). (A) Dynein domains include the positions of mouse and human mutations as well as the buttress (light blue), stalk + MT binding domains (MTBD) (green), 6 AAA ATPase domains (blue), and intra-dynein complex binding domains (turquoise). Human mutations are grouped by color to indicate possible phenotypic similarities (yellow has only motor deficits, while all others have motor deficits and brain malformations [green, blue, red], and intellectual deficits [green, orange, blue, red], and epilepsy [green]). (B) Dynactin p150Glued domains include the coiled-coil (CC) and dynein and MT binding domains and the positions of human mutations (all in MT binding domain). Adapted from reference 3 (A) or 20 (B). Scales indicate amino acid number.
Neurodegenerative Diseases
Researchers have examined the role dynein has in neurodegenerative diseases of the basal ganglia, dementias such as Alzheimer’s disease (AD), and diseases of motor neurons that include amyotrophic lateral scherosis (ALS), spinal muscular atrophy (SMA), and spino-bulbar muscular atrophy (SBMA)2.
Research has identified at least three dynein mutants (Legs at odd angles [Loa], Cramping1, and Sprawling) that affect dynein’s function, leading to motor and/or sensory neuron loss and impaired motor behavior in mice expressing these dynein mutants3,10,12-14 (Fig. 1A). Of these mutants, the Loa mutation (F580Y point mutation in dynein heavy chain) alters dynein’s affinity for dynactin as well as the expression, assembly, and interaction of the protein subunits that compose the dynein complex15. The Loa mutation has been implicated in ALS with reports that mice modeling this disease via overexpression of the Cu/Zn superoxide dismutase enzyme (SOD1) have delayed motor neuron death and an extended lifespan when crossed with Loa+ mice16. In addition, the Loa mutation rescued the axonal transport dysfunction in SOD1 mutant mice3. The mechanism(s) mediating Loa’s amelioration of the ALS phenotype remains unknown with multiple hypotheses being explored, including reduced retrograde transport of cell stress-associated proteins, altered expression of kinesin-1, altered mitochondrial activity and morphology, and/or changes in tau expression 3. Similar to Loa, Cramping1, a point mutation in the dynein heavy chain (Y1055C) which results in abnormalities in motor behavior as well as fat metabolism and storage, has been linked to Huntington’s disease using an in vivo mouse model13. In addition, this dynein mutation induces striatal atrophy in vivo and alters the morphology of neuritic processes in vitro13.
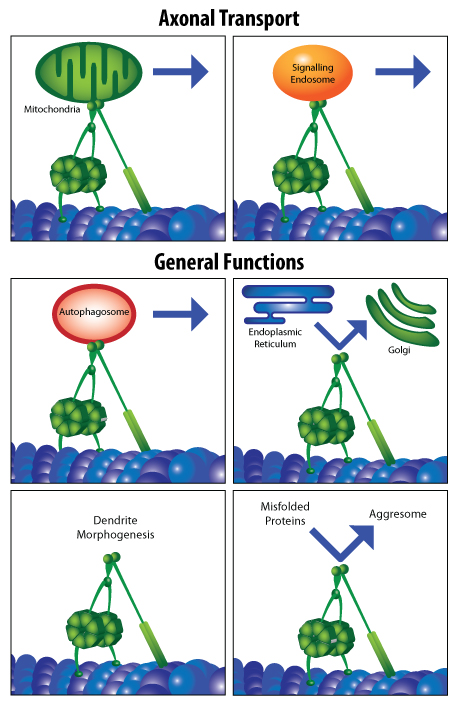
Figure 2: Assorted functions of dynein in the axon and cell body of neurons. Adapted from reference 2.
AD is another neurodegenerative disorder that dynein has been implicated in by virtue of dynein’s role in endocytic pathology. Endocytic dysfunction is considered a marker of early stage AD pathology17,18. RNA interference-induced dynein dysfunction in cell culture mimicked endocytic pathology seen in aged brains of monkeys. Moreover, knockdown of dynein resulted in the accumulation of amyloid precursor protein and its cleavage products, including amyloid beta protein, the primary component of senile plaques19. Thus, dynein-mediated changes in endocytosis appear to play a key role in early AD pathology.
Conclusions
Although the various studies cited above strongly implicate dynein dysfunction in the pathophysiology of several neurodegenerative diseases, its exact role remains unresolved given the indirect (i.e., correlational) nature of most of the studies. Thus, additional research into the role of dynein and its associated protein complexes are necessary to unravel the role this retrograde motor has in normal and disease functions in the central nervous system. At Cytoskeleton, we offer custom-purified cytoplasmic dynein and are developing dynein ATPase assays that can be used to identify inhibitors and enhancers of dynein function. Contact us at tservice@cytoskeleton.com for more information about any of our dynein products.
Related Research Tools and Services
| New proteins made to order... | Application | Module # | Amount | |||||
Dynein (cytoplasmic) | Microtubule stimulated ATPase assay | CS-DN01 | 1 x 50 µg | |||||
MKLP2 motor domain | Microtubule stimulated ATPase assay | 1 x 50 µg | ||||||
KIF7 motor domain | Microtubule stimulated ATPase assay | CS-KF51 | 1 x 100 µg | |||||
Myosin S1 fragment (cardiac) | Actin or thin filament stimulated ATPase assay | CS-MYS03 | 1 x 250 µg | |||||
Myosin S1 fragment (skeletal) | F-actin stimulated ATPase assay | CS-MYS04 | 1 x 250 µg | |||||
Myosin S1 fragment (smooth) | F-actin stimulated ATPase assay | CS-MYS05 | 1 x 250 µg | |||||
Myosin S1 fragment (non-muscle) | F-actin stimulated ATPase assay | CS-MYS06 | 1 x 250 µg | |||||
Actin Thin Filaments | Calcium sensitive F-actin stimulated myosin ATPase assay | CS-TFC01 | 1 x 1 mg | |||||
Tropomyosin / Tropomodulin | Actin thin filament preparation | CS-TT05 | 1 x 1 mg | |||||
| Kinesin and Dynein Proteins | Cat. # | Amount | ||||||
CENP-E Motor Domain Protein | CP01-A | 2 x 25 µg | ||||||
Chromokinesin Motor Domain Protein | CR01-A | 2 x 25 µg | ||||||
Eg5 Motor Domain Protein | EG01-A | 2 x 25 µg | ||||||
Eg5 Homolog BimC Motor Domain Protein | BM01-A | 2 x 25 µg | ||||||
Eg5 Homolog BimC Motor Domain Protein | EG02-A | 2 x 15 µg | ||||||
KIFC3 Motor Domain Protein | KC01-A | 2 x 25 µg | ||||||
KIF3C Motor Domain Protein | KF01-A | 2 x 25 µg | ||||||
Kinesin Heavy Chain Motor Domain Protein | KR01-A | 2 x 25 µg | ||||||
MCAK Motor Domain Protein | MK01-A | 2 x 25 µg | ||||||
MKLP1 Motor Domain Protein | MP01-A | 2 x 25 µg | ||||||
Kinesin ELIPA Biochem Kit | BK060 | 96 assays | ||||||
Kinesin ATPase Endpoint Assay | BK053 | 1000 assays | ||||||
Microtubules (pre-formed) | MT002 | 4 x 500 µg | ||||||
Actin Filaments (pre-formed) | AKF99 | 1 x 1 mg | ||||||
References
- Allan V.J. 2011. Cytoplasmic dynein. Biochem. Soc. Trans. 39, 1169-1178.
- Eschbach J. & Dupuis L. 2011. Cytoplasmic dynein in neurodegeneration. Pharmacol. Ther. 130, 348-363.
- Schiavo G. et al. 2013. Cytoplasmic dynein heavy chain: the servant of many masters. Trends Neurosci. 36, 641-651.
- Puls I. et al. 2003. Mutant dynactin in motor neuron disease. Nat. Genet. 33, 455-456.
- Farrer M.J., et al. 2009. DCTN1 mutations in Perry syndrome. Nat. Genet. 41, 163-165.
- Weedon M.N. et al. 2011. Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am. J. Hum. Genet. 89, 308-312.
- Harms M.B. et al. 2012. Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology. 78, 1714-1720.
- Willemsen M.H. et al. 2012. Mutations in DYNC1H1 cause severe intellectual disability with neuronal migration defects. J. Med. Genet. 49, 179-183.
- LaMonte B.H. et al. 2002. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron. 34, 715-727.
- Hafezparast M. et al. 2003. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science. 300, 808-812.
- Kimura N. et al. 2012. Dynein dysfunction disrupts intracellular vesicle trafficking bidirectionally and perturbs synaptic vesicle docking via endocytic disturbances. Am. J. Pathol. 180, 550-561.
- Chen X-J. et al. 2007. Proprioceptive sensory neuropathy in mice with a mutation in the cytoplasmic dynein heavy chain 1 gene. J. Neurosci. 27, 14515-14524.
- Braunstein K.E. et al. 2010. A point mutation in the dynein heavy chain gene leads to striatal atrophy and compromises neurite outgrowth of striatal neurons. Hum. Mol. Genet. 19, 4385-4398.
- Ori-McKenney K.M. et al. 2010. A cytoplasmic dynein tail mutation impairs motor processivity. Nat. Cell Biol. 12, 1228-1234.
- Deng W. et al. 2010. Neurodegenerative mutation in cytoplasmic dynein alters its organization and dynein-dynactin and dynein-kinesin interactions. J. Biol. Chem. 285, 39922-39934.
- Kieran D. et al. 2005. A mutation in dynein rescues axonal transport defects and extends the life span of ALS mice. J. Cell Biol. 169, 561-567.
- Cataldo A.M. et al. 2004. Abeta localization in abnormal endosomes: association with earliest Abeta elevations in AD and Down syndrome. Neurobiol. Aging. 25, 1263-1272.
- Nixon R.A. 2005. Endosome function and dysfunction in Alzheimer's disease and other neurodegenerative diseases. Neurobiol. Aging. 26, 373-382.
- Kimura N. et al. 2009. Dynein dysfunction induces endocytic pathology accompanied by an increase in Rab GTPases. J. Biol. Chem. 284, 31291-31302.
- Vallee R.B. et al. 2012. Multiple modes of cytoplasmic dynein regulation. Nat. Cell Biol. 14, 224-230.