May Newsletter : Microglia and Neurodegenerative Diseases
Microglia are the primary immune cells, the so-called professional phagocytes, of the mammalian brain. Microglia continually scan the entire brain in their resting state and when neurotoxic pathogens and damaged cellular machinery are detected, they are activated to eliminate them1,2 (Fig. 1). In healthy neurons, microglia also contribute to essential cellular functions such as neurogenesis, neurodevelopment, and neural plasticity3,4. Of similar importance are the roles microglia have in neurodegenerative diseases characterized by aggregates of pathogenic, misfolded proteins such as Parkinson’s disease (PD; Lewy bodies) and Alzheimer’s disease (AD; Aβ plaques). Microglia-mediated neuroinflammation is a common pathophysiology in PD and AD human brains, as well as in in vivo animal model brains1,2,4-9. Moreover, recent genetic and transcriptomic studies revealed microglia-associated signaling cascades as critical players in AD pathogenesis1,7. This newsletter discusses microglia activation and neuroinflammation in PD and AD.
PD is characterized by a loss of dopamine (DA) neurons in the substantia nigra pars compacta, a corresponding loss of dopaminergic (DAergic) terminals throughout the basal ganglia, and Lewy bodies which are composed primarily of intracellular α-synuclein aggregates. Lewy bodies are a pathophysiological hallmark of PD and trigger microglia-mediated neuroinflammation, which itself is another neuropathological correlate of PD1,2,8,10 (Fig. 1). In the brains of PD patients, Lewy body neurites are associated with activated microglia and α-synuclein deposits are correlated with inflammatory markers. In vitro cell culture and in vivo animal models also demonstrate a relationship between α-synuclein aggregates and microglial activation2,8,10. In vitro, α-synuclein concentration-dependently activates microglia and microglia can also remove α-synuclein aggregates through phagocytosis, though the exact relationship between the two processes remains unclarified10. Over-expressing wild-type or mutant α-synuclein in mice produces early microglial activation.

In vivo over-expression of wild-type human α-synuclein in mouse PD models display early and sustained microglial activation and increased release of multiple pro-inflammatory cytokines (e.g., tumor necrosis factor-alpha [TNF-α], interleukins [IL] such as IL-1b, IL-6, and IL-18) and chemokines2,6. This inflammatory response is restricted to midbrain DA neurons that comprise the nigrostriatal pathway, even though α-synuclein expression encompasses the entire brain2,6,8. In the MPTP mouse model of PD, the MPTP toxin provokes microglial-mediated inflammation with the expected increase in pro-inflammatory cytokines prior to MPTP-induced DAergic neuron neurodegeneration
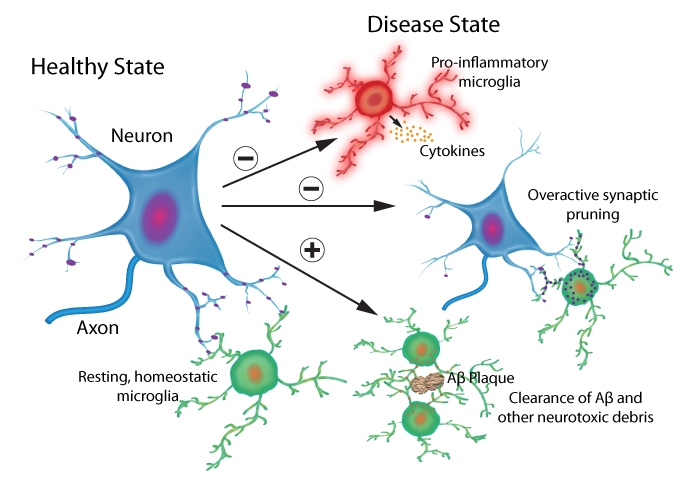
Fig. 1. Physiological and pathological microglial processes. Microglia-mediated signaling cascades trigger neuroprotective/neurotrophic and neurotoxic effects on healthy and diseased neurons.
The classic histopathological hallmarks of AD are extracellular Aβ plaque deposits and intracellular hyperphosphorylated tau tangles. Microglia activation and release of pro-inflammatory cytokines, and the subsequent neuroinflammation are also pathological hallmarks of AD1,2,7 (Fig. 1). Studies support neuroprotective and neurotoxic roles for microglia and associated proteins in AD. On the one hand, pathogenic Aβ soluble species and insoluble plaques undergo phagocytosis, and at later stages of AD, microglia encircle and compact plaques (Fig. 1). Compaction may prevent further deposition onto the existing plaque1,2,7. Additionally, recent genetic studies reveal multiple AD risk genes, many of which are related to microglia either directly or indirectly (e.g., triggering receptor expressed on myeloid cells 2 [TREM2] and complement receptor 1 [CR1] genes)1,7. TREM2 is necessary for phagocytic signaling and microglia-mediated neuroprotection, and its dysfunction exacerbates AD pathophysiology1,2,7. In healthy neurons, CR1 and other members of the innate immune system’s complement signaling cascade assists microglia in phagocytosis, removal of immune complexes, and negative regulation of the complement system11,12, as well as the removal of excess synapses (i.e., synaptic pruning) during development, maturation, and refinement of neuronal circuitry13-15. Neurotoxicity occurs when the complement proteins are up-regulated and activated in human AD brains and in mouse AD models13-16. Interestingly, the up-regulation occurs prior to Aβ plaque deposition and when microglial dysfunction is observed13-16. These observations suggest that dysfunctional microglia and complement proteins contribute to early synapse loss due to a malfunctioning of the physiological synaptic pruning process, resulting in microglia engulfing mature, functional synapses1,2,16 (Fig. 1). AD-associated synapse loss and synaptic dysfunction are rescued following inhibition of complement signaling1,16. These data are noteworthy because synapse loss is an early pathological event in AD (and similar diseases) and robustly correlates with cognitive decline17,18. Microglia dysfunction is exacerbated with continuing plaque formation as in vivo two-photon microscopy in different AD mouse models shows that as Aβ plaque numbers increase, microglia function decreases19.
Microglia and microglia-associated proteins are implicated in tau pathology. Through an unknown pathway, activation of complement proteins appears to worsen tau pathology in in vivo mouse models20,21. Moreover, over-activation of microglia in an in vivo transgenic tau mouse model worsened the onset and progression of tau pathology22,23. An in vivo model of mutant tau pathology suggests that microglial uptake of toxic tau species and exosomal release of tau contributes to cell-to-cell transmission of the pathogenic tau7,24.
Summary
Microglia display a diverse set of physiological and pathological functions in the brain which presents multiple challenges. In neurodegenerative diseases, an open question is when and how the switch from anti- to pro-inflammatory signaling occurs. Understanding the sequence and timing of alterations in the two opposing microglial phenotypes is a complicated undertaking which requires new research tools25. Indeed, a recent advance in live cell imaging with calcium indicators offers an opportunity to better understand the in vivo role of microglia in neuron function and dysfunction26. Cytoskeleton, Inc. offers a variety of tools, including live cell imaging reagents, purified cytoskeletal proteins, functional assay kits, and Signal-Seeker™ kits for quantifying post-translational modifications of target proteins to decipher microglia-regulated signaling cascades in the mammalian brain.
References
-
Salter M.W. and Stevens B. 2017. Microglia emerge as central players in brain disease. Nat. Med. 23, 1018-1027
-
Wolf S.A. et al. 2017. Microglia in physiology and disease. Annu. Rev. Physiol. 79, 619–643.
-
Bilimoria P.M. and Stevens B. 2015. Microglia function during brain development: New insights from animal models. Brain Res. 1617, 7-17.
-
Sominsky L. et al. 2018. Microglia: Key players in neurodevelopment and neuronal plasticity. Int. J. Biochem. Cell Biol. 94, 56-60.
-
Shen Z. et al. 2017. Microglia-targeted stem cell therapies for Alzheimer disease: a preclinical data review. J. Neurosci. Res. 95, 2420-2429.
-
Lee S.-H. and Suk K. 2018. Kinase-based taming of brain microglia toward disease-modifying therapy. Front. Cell. Neurosci. 12, 474.
-
Hansen D.V. et al. 2018. Microglia in Alzheimer’s disease. J. Cell Biol. 217, 459-472.
-
Subramaniam S. and Federoff J. 2017. Targeting microglial activation states as a therapeutic avenue in Parkinson’s disease. Front. Aging Neurosci. 9, 176.
-
Ransohoff R.M. 2016. How neuroinflammation contributes to neurodegeneration. Science. 353, 777-783.
-
Zhang Q.S. et al. 2017. Pathological alpha-synuclein exacerbates the progression of Parkinson’s disease through microglial activation. Toxicol. Lett. 265, 30-37.
-
Khera R. and Das N. 2009. Complement Receptor 1: disease associations and therapeutic implications. Mol. Immunol. 46, 761-772.
-
Fonseca M.I. et al. 2016. Analysis of the putative role of CR1 in Alzheimer’s disease: genetic association, expression, and function. PLoS ONE 11, e0149792.
-
Schafer D.P. et al. 2012. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 74, 691-705.
-
Stephan A.H. et al. 2012. The complement system: an unexpected role in synaptic pruning during development and disease. Annu. Rev. Neurosci. 35, 369-389.
-
Stevens B. et al. 2007. The classical complement cascade mediates CNS synapse elimination. Cell. 131, 1164-1178.
-
Hong S. et al. 2016. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 352, 712-716.
-
Terry R.D. et al. 1991. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572-580.
-
DeKosky S.T. et al. 1996. Structural correlates of cognition in dementia: quantification and assessment of synaptic change. Neurodegeneration. 5, 417-421.
-
Krabbe G. et al. 2013. Functional impairment of microglia coincides with beta-amyloid deposition in mice with Alzheimer-like pathology. PloS ONE. 8, e60921.
-
Fonseca M.I. et al. 2009. Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer’s disease. J. Immunol. 183, 1375-1383.
-
Britschgi M. et al. 2012. Deficiency of terminal complement pathway inhibitor promotes neuronal tau pathology and degeneration in mice. J. Neuroinflammation. 9, 220.
-
Bhaskar K. et al. 2010. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 68, 19-31.
-
Maphis N. et al. 2015. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain. 138, 1738-1755.
-
Asai H. et al. 2015. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 18, 1584-1593.
-
Tang Y. and Le W. 2015. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 53, 1181-1194.
-
Brawek B. et al. 2017. A new approach for ratiometric in vivo calcium imaging of microglia. Sci. Rep. 7, 6030.
Related Products
Actin Products
Actin protein (>99% pure): bovine cardiac muscle (Cat. # AD99)
Actin protein (>99% pure): chicken gizzard muscle (Cat. # AS99)
Actin protein (pre-formed filaments): rabbit skeletal muscle (Cat. # AKF99)
Actin protein (>95% pure): rabbit skeletal muscle (Cat. # AKL95)
Actin protein (>99% pure): rabbit skeletal muscle (Cat. # AKL99)
Actin protein (>99% pure): human platelet (Cat. # APHL99)
Actin protein (rhodamine): human platelet (Cat. # APHR)
Actin protein (rhodamine): rabbit skeletal muscle (Cat. # AR05)
Spirochrome SiR-Actin Kit (Cat. # CY-SC001)
Spirochrome SiR700-Actin Kit (Cat. # CY-SC013)
Acti-stain 488 Phalloidin (Cat. # PHDG1)
Acti-stain 555 Phalloidin (Cat. # PHDH1)
Acti-stain 670 Phalloidin (Cat. # PHDN1)
Rhodamine Phalloidin (Cat. # PHDR1)
Actin Biochem Kits
Actin Binding Protein Spin-Down Assay Biochem Kit: rabbit skeletal muscle actin (Cat. # BK001)
Actin Binding Protein Spin-Down Assay Biochem Kit: human platelet actin (Cat. # BK013)
Actin Polymerization Biochem Kit (fluorescence format): rabbit skeletal muscle actin (Cat. # BK003)