August Newsletter: Rab GTPases and Neurodegeneration
Rab GTPases, members of the Ras super-family of GTPases, express at least 60 isoforms in humans1 with 24 enriched in, or specific for, the central nervous system (CNS)2. Rab GTPases are integral regulators of intracellular membrane trafficking, regulating the formation, maturation, transport, tethering, and fusion of vesicles in the endomembrane system3. In neurons, Rab-mediated membrane/vesicle trafficking is involved in virtually every aspect of neuron physiology, and dysfunction has been implicated in several neurodegenerative diseases2,4-7 (Fig. 1). Indeed, dysfunctional membrane trafficking is an early marker of neurodegeneration5. Here, Rab-mediated trafficking defects in Parkinson’s disease (PD) and Alzheimer’s disease (AD) are reviewed.
The cause of PD is primarily idiopathic – only a small fraction of PD cases are familial and attributable to mutations in proteins that also are involved in Rab activity and membrane trafficking. Mutant proteins include Rab39b, α-synuclein, PTEN-induced putative kinase (PINK1), and leucine-rich repeat kinase 2 (LRRK2)4,6,7. Rab39b is particularly interesting because loss-of-function mutations in this gene are directly linked to inherited early-onset PD with Lewy body pathology and also symptoms of intellectual disability8-10. Rab39b is selectively expressed in neurons and regulates AMPA receptor (AMPAR) subunit GluA2 trafficking to the post-synaptic membranes of hippocampal neurons4. Rab39b mutations result in the formation of AMPARs devoid of GluA211,12, which are associated with immature synapses and intellectual disability; however, how these AMPARs contribute to the selective degeneration of dopaminergic neurons remains unknown4. α-synuclein is the primary component of intracellular protein aggregates which make up Lewy bodies, one of the pathological hallmarks of PD5,6,12. α-synuclein interacts and colocalizes with multiple Rab isoforms, including Rab1, 3a, 5, 7, 8, 11, 13, and 35, for the purpose of membrane trafficking regulation (Fig. 1).

Overexpression of certain Rabs (Rab1, 3a, 8, 11, 13) reverses some or all of the mutant or overexpressed wild-type α-synuclein-induced deficits in ER/Golgi trafficking, locomotor activity, and dopamine neuron survival in<span> </span><em>in vitro</em><span> </span>and<span> </span><em>in vivo</em><span> </span>PD models<sup>4,6,12</sup>. Conversely, overexpression of Rab35 increased mutant α-synuclein aggregation in dopamine neurons<sup>4</sup>. Rab8a, 8b, and 13 are phosphorylated following PINK1 activation but not by PINK1 itself<sup>13</sup>. Rab8 phosphorylation is prevented by PINK1 knockdown or mutation in cell lines<sup>13</sup>. This suggests that Rab8-mediated post-Golgi protein trafficking may be negatively impacted by PINK1 mutants in PD patients<sup>4</sup>. LRRK2 is a mammalian kinase involved in synaptic vesicle recycling and autophagy<sup>14</sup>. Lrrk, the Drosophila homolog of LRRK2, interacts directly with, and regulates, Rab7, an isoform associated with late endosomes/lysosomes<sup>15</sup>. Constitutively-active LRRK2 mutants inhibit Rab7-mediated protein trafficking from early to late endosomes, and overexpression of Rab7 rescues this suppression<sup>16</sup>. Other Rab isoforms interacting with LRRK2 are Rab3, 5, 8, 10, 12, 32, 35, and 38<sup>4,6,7,17</sup>. Taken together, these studies strongly suggest that Rab GTPases contribute to PD pathogenesis, often through indirect relationships with pathogenic mutant proteins that disrupt the homeostasis of Rab-mediated membrane/vesicle trafficking (Fig. 1). Interesting questions remain, including if a single Rab isoform exists whose dysfunction is directly linked to idiopathic PD or if pathogenesis results from a general, widespread dysfunction in Rab-mediated membrane trafficking<sup>7</sup>?</p>
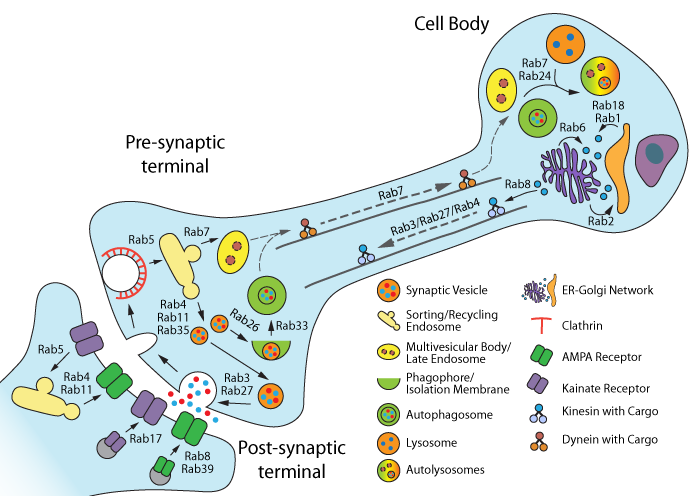
Figure 1. Rab GTPase functions in neurons. Figure adapted from reference # 4.
Similar to PD, most cases of AD are idiopathic. Determining the contribution of defective Rab-mediated membrane trafficking to onset of cognitive impairment and eventual pathophysiology of amyloid beta (Ab) plaques and neurofibrillary tangles is a work in progress. Importantly, endolysosomal deficiencies precede deposition of Ab in idiopathic AD. Current thinking is that there is an imbalance in Rab GTPase expression levels and/or activity that occurs early in AD pathogenesis. Early endolysosomal deficiencies include enlarged Rab5-positive early endosomes and lysosome accumulation4 and upregulation of Rab4, 5, 7, and 27 mRNA and protein levels in post-mortem cholinergic basal forebrain and hippocampal neurons of patients diagnosed with either mild cognitive impairment or AD18,19. These findings suggest that Rab-mediated endocytic pathways are over-activated in AD, which then in turn negatively affect processing/trafficking of proteins, including Ab and tau.
For familial AD which is linked to mutations in presenilin 1 (PSEN1), presenilin 2 (PSEN2), and amyloid precursor protein (APP), similar mechanistic changes in Rab-mediated endolysosomal signaling are likely4,5. Presenilins are proteases involved in the processing of APP, the precursor for pathological Ab species, protein trafficking and turnover, and autophagosome/lysosome functions4,5. Functional PSEN1 is required for proper Rab6 activation which is achieved through Rab6 membrane localization. Accordingly, PSEN1 loss of fuction impairs Rab6-mediated protein trafficking between the Golgi and ER20. Reduced levels of active Rab GTPases may trigger a hyper-compensatory response which further exacerbates the imbalance in Rab GTPase expression and/or function. For example, in AD patients, Rab6 expression levels are increased 5-fold, while in PSEN1 mutant cells, Rab4 and 6 expression levels are increased4. Conversely, cells expressing mutant PSEN1 displayed reduced levels of Rab8-associated membranes4. Rab GTPases might also work in concert with PSEN2 as this presenilin isoform is specifically localized to late endosomes/lysosomes where it cleaves proteins trafficked to these compartments4. In this capacity, mutant PSEN2 results in increased levels of pathological Ab in these compartments, which could compromise endosomal/lysosomal function and provide another avenue by which Rabs contribute to AD pathogenesis.
Summary
In PD and AD, Rab GTPases have altered activity, regulation, and/or expression. Perhaps the clearest lesson from studies of Rabs and neurodegeneration is that maintaining a balance (i.e., homeostasis) in Rab-mediated activity, localization, and expression levels are essential as certain Rab isoforms can ameliorate the deleterious effects of pathological mutant proteins in PD and AD, while the opposite effect can occur with other Rab isoforms. Rab homeostasis is key to preventing early pathogenic changes in the endolysosomal system. Notably, the phosphorylation status of Rab GTPases is also of interest given the direct and indirect interactions with multiple kinases. Cytoskeleton offers Signal-Seeker™ Detection Kits to measure endogenous levels of various post-translational modifications (PTMs), including tyrosine phosphorylation, acetylation, ubiquitination, and SUMOylation, allowing the researcher to assemble a PTM profile for the experimental proteins of interest.
Related Products
Signal Seeker™ Kits
New Signal-Seeker™ Acetyl-Lysine Detection Kit (30 assay) (Cat. # BK163)
New Signal-Seeker™ Acetyl-Lysine Detection Kit (10 assay) (Cat. # BK163-S)
Signal-Seeker™ Phosphotyrosine Detection Kit (30 assay) (Cat. # BK160)
Signal-Seeker™ Phosphotyrosine Detection Kit (10 assay) (Cat. # BK160-S)
Signal-Seeker™ Ubiquitination Detection Kit (30 assay) (Cat. # BK161)
Signal-Seeker™ Ubiquitination Detection Kit (10 assay) (Cat. # BK161-S)
Signal-Seeker™ SUMOylation 2/3 Detection Kit (30 assay) (Cat. # BK162)
Signal-Seeker™ SUMOylation 2/3 Detection Kit (10 assay) (Cat. # BK162-S)
PTM Antibodies, Beads, Etc
New Acetyl-Lysine Antibody Mouse Monoclonal (7B5A1) (Cat. # AAC02)
New Acetyl-Lysine-HRP Antibody Mouse Monoclonal (19C4B2.1) (Cat. # AAC03-HRP)
New Acetyl-Lysine Affinity Beads (Cat. # AAC04-beads)
Phosphotyrosine-HRP Mouse Monoclonal Antibody 27B10 (Cat. # APY03-HRP)
SUMOylation 2/3 Affinity Beads (Cat. # ASM24-beads)
Ubiquitin Antibody Mouse Monoclonal (Cat. # AUB01)
References
Brighouse A. et al. 2010. Rab protein evolution and the history of the eukaryotic endomembrane system. Cell. Mol. Life Sci. 67, 3449–3465.
D’Adamo P. et al. 2014. RAB GTPases and RAB-interacting proteins and their role in the control of cognitive functions. Neurosci. Biobehav. Rev. 46, 302-314.
Binotti B. et al. 2016. Functions of Rab proteins at presynaptic sites. Cells. 5, E7.
Kiral F.R. et al. 2018. Rab GTPases and membrane trafficking in neurodegeneration. Curr. Biol. 28, R471-R486.
Wang D. et al. 2013. Membrane trafficking in neuronal maintenance and degeneration. Cell. Mol. Life Sci. 70, 2919-2934.
Shi M.-M. et al. 2017. Rab GTPases: The key players in the molecular pathway of Parkinson’s disease. Front. Cell. Neurosci. 11, 81.
Clague M.J. and Rochin L. 2016. Parkinson’s disease: A traffic jam? Curr. Biol. 26, R332-R334.
Wilson G.R. et al. 2014. Mutations in RAB39B cause X-linked intellectual disability and early-onset Parkinson disease with α-synuclein pathology. Am. J. Hum. Genet. 95, 729-735.
Lesage S. et al. 2015. Loss-of-function mutations in RAB39B are associated with typical early-onset Parkinson disease. Neurol. Genet. 1, e9.
Mata I.F. et al. 2015. The RAB39B p.G192R mutation causes X-linked dominant Parkinson’s disease. Mol. Neurodegener. 10, 50.
Mignogna M.L. et al. 2015. The intellectual disability protein RAB39B selectively regulates GluA2 trafficking to determine synaptic AMPAR composition. Nat. Commun. 6, 6504.
Tang B.L. 2017. Rabs, membrane dynamics, and Parkinson’s disease. J. Cell Physiol. 232, 1626-1633.
Lai Y.-C. et al. 2015. Phosphoproteomic screening identifies Rab GTPases as novel downstream targets of PINK1. EMBO J. 34, 2840-2861.
Shin N. et al. 2008. LRRK2 regulates synaptic vesicle endocytosis. Exp. Cell Res. 314, 2055-2065.
Dodson M.W. et al. 2012. Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum. Mol. Genet. 21, 1350-1363.
Gomez-Suaga P. 2014. LRRK2 delays degradative receptor trafficking by impeding late endosomal budding through decreasing Rab7 activity. Hum. Mol. Genet. 23, 6779-6796.
Steger M. et al. 2017. Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. eLife. 6, e31012.
Ginsberg S.D. et al. 2010. Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer’s disease progression. Biol. Psychiatry. 68, 885-893.
Ginsberg S.D. et al. 2011. Upregulation of select rab GTPases in cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer’s disease. J. Chem. Neuroanat. 42, 102-110.
Scheper W. et al. 2004. Rab6 membrane association is dependent of Presenilin 1 and cellular phosphorylation events. Mol. Brain Res. 122, 17-23.