Ras Cancer Therapeutics: 5 Promising Targets
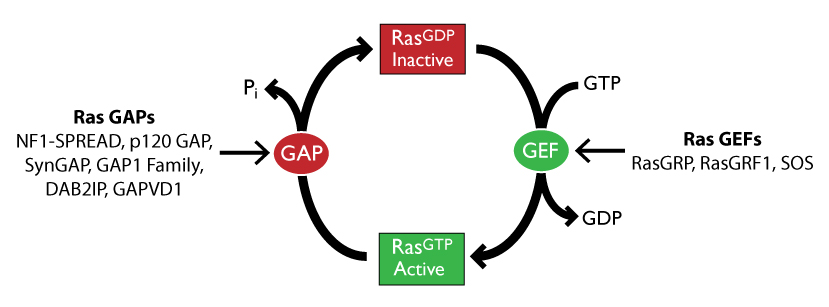
The Ras GTPase plays an important role in multiple signal transduction pathways involved in normal cell growth and differentiation as well as several forms of cancer1,2. The three isoforms of Ras, H-Ras, N-Ras, and K-Ras, were identified over 30 years ago for their oncogenic activation in human tumors1,2. Aberrant Ras signaling has been identified in more than 30% of all human cancers with the most common being lung, colon, and pancreatic cancers1,3. In particular, K-Ras has been identified as the most important Ras protein in cancer research, correlated with over 21% of human cancers4. Despite extensive research on these proteins, no effective Ras inhibitor has been identified, earning K-Ras the reputation of an undruggable protein. Many efforts in the past have been unsuccessful, but recent advances in the field have lead to new hope in developing a direct therapeutic inhibitor to Ras protein as well as indirect inhibitors targeting GEFs, GAPs, downstream proteins, and post-translational modifications (Fig. 1). Here we briefly highlight several of the targets that show promise for Ras cancer therapeutics.

1. Targeting Mutant K-Ras Directly
Mutant vs wildtype K-Ras selectivity has proved challenging since mutations are distal from the effector binding sites1. The recent discovery of an allosteric switch II pocket (S-IIP) in a K-Ras mutant (G12C) provided a new target for K-Ras inhibition5. Soon after, the development of small molecules that bind to this mutant cysteine were found to disrupt SOS-mediated nucleotide exchange, thus inhibiting mutant K-Ras and giving new hope to direct K-Ras therapies via small molecules5. To read more regarding SOS and Ras inhibition, please refer to the July 2014 newsletter "New small molecule inhibitors to control Ras signaling via Sos/K-Ras binding" found here: www.cytoskeleton.com/blog/kras-sos-inhibitor-news.
2. Targeting GEFs and GAPs
SOS, RasGRF, and RasGRP are the three classes of GEFs that mammals express that are typically regarded as the activators of Ras6. Despite GEF-mediated Ras activation, mutations in GEFs are surprisingly uncommon in cancer. On the other hand, GAPs, including classes NF1, p120GAP/RASA1, SynGAP/RASA5, GAP1 family, DAB2IP, and GAPVD1, have been associated with several Ras-linked cancers6,7,8 (Fig. 1). For more about the role of GEFs/GAPs in tumorigenesis, please refer to the recent review by Dr. Anne Hennig et al6.
3. Targeting Downstream Proteins
Mitogen-activated protein kinase (MAPK) kinase (MEK), a protein in the MAPK pathway downstream of Ras activation, is targeted by the FDA approved drug trametinib (MekinistTM), which has been shown to be clinically active in some patients9. However, maintaining the necessary amount of active drug in the bloodstream without serious side effects has been challenging9. Another pathway downstream of Ras activation is the Merlin/Hippo protein kinase pathway which regulates the activity of the pro-growth transcriptional co-activator YAP. Activation of this pathway restricts cell proliferation and promotes apoptosis and this pathway is critical for Ras-dependent cancers10-12. Merlin regulates the Hippo pathway as its loss results in inactivation of the Hippo pathway10. The site of Merlin's suppression of tumorigenesis remains unclear with papers supporting both a membrane and nuclear localization. However, a recent paper suggests that rather than suppressing tumorigenesis via the Hippo pathway at the plasma membrane, Merlin acts by inhibiting the E3 ubiquitin ligase complex CRL4DCAF1 interaction with Hippo pathway serine/threonine kinases LATS1/2 in the nucleus13. Nuclear-localized Merlin interacts with CRL4DCAF1, preventing this complex from ubiquitinating LATS kinases which can then phosphorylate YAP, inactivating it and triggering YAP's accumulation in the cytoplasm10. However, if YAP is active (unphosphorylated), it partners with TEAD transcription factors to increase expression of mutant and wild-type Ras proteins10,13. A more detailed review of YAP1 and the Hippo Pathway can be found in our April 2015 newsletter found here: www.cytoskeleton.com/yap1-ras-oncogenic-review-detail
4. Targeting Post-translational Modifications
Ras proteins undergo several post-translational modifications, including proteolytic cleavage by RCE1, farnesylation, carboxymethylation by ICMT, and palmitoylation14. Recently, additional post-translational modifications such as mono-ubiquitination and acetylation have been shown to play a key role in regulating Ras activation. Mono-ubiquitination occurs at K-Ras Lys147 which enhances GTP loading15. Acetylation at K-Ras Lys104 decreases GEF-induced nucleotide exchange, suggesting SIRT2 and HDAC6 deacetylases as potential targets to inhibit16.
5. Nanomedicine and siRNA Knockdown
A recent study suggests that targeting mutant K-Ras via combined delivery of siRNA and arsenic-encapsulated vesicles to induce apoptosis and down-regulation of the mutant K-Ras's ability to proliferate and migrate in pancreatic cancer treatment is worthwhile17. Moreover, siRNA treatments targeting the inhibition of K-Ras in combination with RAF and PI3K can impair mutant colorectal cancer in xenograft models18.
Summary
As researchers discover additional Ras regulatory mechanisms, sensitive and quantitative methods to measure activated levels of this GTPase take on increasing importance. To aid researchers with identifying and characterizing cancer therapeutics, Cytoskeleton, Inc. offers many small G-protein tools such as pull-down and G-LISA activation assays which are widely used to measure the activated levels of small G-proteins. Small G-protein antibodies, activators, and inhibitors are also available to help elucidate Ras activation pathways.
Related Products & Resources
Anti-Acetyl Lysine Mouse Monoclonal Antibody (Cat. # AAC01)
NEW! Anti-Phosphotyrosine Mouse Monoclonal Antibody (Cat. # APY03)
NEW! Anti-SUMO 2/3 Mouse Monoclonal Antibody (Cat. # ASM23)
Anti-Ubiquitin Mouse Monoclonal Antibody (Cat. # AUB01)
Ras G-LISA Activation Assay (Cat. # BK131)
Ras Pull-down Activation Assay (Cat. # BK008)
RhoGEF Exchange Assay Biochem Kit (in vitro fluorescence format) (Cat. # BK100)
SOS exchange domain (564-1049) protein (Cat. # CS-SOS1)
Activation Assay Video
 Cytoskeleton's Small G-protein Activation Assays come in a traditional pull-down bead format or our advanced ELISA based G-LISA format. Learn which Activation Assay format is right for you by watching this video.
Cytoskeleton's Small G-protein Activation Assays come in a traditional pull-down bead format or our advanced ELISA based G-LISA format. Learn which Activation Assay format is right for you by watching this video.
References
- Wang W. et al. 2012. Ras inhibition via direct Ras binding—is there a path forward. Bioorg. Med. Chem. Lett. 22, 5766-5776.
- Castellano E. and Santos E. 2011. Functional specificity of Ras isoform: so similar but so different. Genes Cancer. 2, 216-231.
- Burns M.C. et al. 2014. Approach for targeting Ras with small molecules that activate SOS-mediated nucleotide exchange. Proc. Natl. Acad. Sci. U.S.A. 111, 3401-3406.
- Baines A. et al. 2011. Inhibition of Ras for cancer treatment: the search continues. Future Med. Chem. 3, 1787-1808.
- Ostrem J. et al. 2013. K-Ras (G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 503, 548-551.
- Hennig A. et al. 2015. Ras activation revisited: role of GEF and GAP systems. Biol. Chem. doi: 10.1515/hsz-2014-0257.
- Nissan M.H. et al. 2014. Loss of NF1 in Cutaneous Melenama Is Associated with RAS Activation and MEK Dependence. Cancer Res. 74, 2340
- Stephen A.G. et al. 2014. Dragging Ras Back in the Ring. Cancer Cell. 25, 272-281.
- Online: http://www.cancer.gov/cancertopics/research-updates/2013/MEK.
- Online: http://www.cancer.gov/researchandfunding/priorities/ras/advance-research/events/ras-fagin-video.Hong X. et al. 2014.
- Opposing activities of the Ras and Hippo pathways converge on regulation of YAP protein turnover. EMBO J. 33, 2447-2457.
- Kapoor A. et al. 2014. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell. 158, 185-197.
- Li W. et al. 2014. Merlin/NF2 loss-driven tumorigenesis linked to CRL4(DCAF1)-mediated inhibition of the hippo pathway kinases Lats1 and 2 in the nucleus. Cancer Cell. 26, 48-60.
- Gysin S. et al. 2011. Therapeutic strategies for targeting ras proteins. Genes Cancer. 2, 359-372.
- Sasaki A.T. et al. 2011. Ubiquitination of K-Ras enhances activation and facilitates binding to select downstream effectors. Sci. Signal. 4, ra13.
- Yang M.H. 2013. HDAC6 and SIRT2 Regulate the acetylation state and oncogenic activity of mutant K-RAS. Mol. Cancer Res. 11, 1072-1077.
- Zeng L. et al. 2014. Combination of siRNA-directed Kras oncogene silencing and arsenic-induced apoptosis using a nanomedicine strategy for the effective treatment of pancreatic cancer. Nanomedicine. 10, 463-472.
- Yuan T.L. et a. 2014. Development of siRNA Payloads to Target KRAS-Mutant Cancer. Cancer Discovery. 4, 1182.