Ras and Rho Post-translational Modification by Prenylation: Role in Cancer Drug Discovery
Ras and Rho GTPases are small G-proteins that cycle between an active GTP-bound form and inactive GDP-bound form. Ras proteins, known for their role in cell proliferation, and Rho proteins, known for their involvement in cell morphology, have common post-translational modifications (PTMs) that have been identified as contributors to oncogenesis1,2. Understanding Ras and Rho PTMs have been of interest for drug discovery groups for many years. Recent studies of signaling pathways mediated by the Ras and Rho PTMs prenylation and/or palmitoylation have identified potential cancer drug targets1,2.
Lipid Modification of Ras: A PTM’s Role in Cancer
Mutations resulting in abnormal activation of Ras isoforms (H, N, & K) are a common cause of human cancers, including pancreatic, cervical, colon, lung, thyroid, bladder, breast, skin, and leukemias1,3,4. Although activated Ras isoforms have been frequently associated with cancers, no effective Ras signaling inhibitor has been developed. While all three isoforms are worthy drug targets1, recent efforts have focused on K-Ras5. (Fig. 1).
K-Ras undergoes the PTM farnesylation (a type of prenylation) that involves adding an isoprenyl group on the C-terminus6,7. In short, either a farnesyl or geranylgeranyl lipid is covalently attached to the cysteine residue on the C termini of Ras proteins (CAAX tetrapeptide motif), followed by Rce1-mediated amino acid cleavage of -AAX, and Icmt-mediated methylation of the prenylated cysteine6,7.
The farnesyl tails of K-Ras tether the protein to cell membranes and help restrict free diffusion of K-Ras through the cytoplasm6-8. These farnesyl tails also play an important role in trafficking Ras to proper subcellular compartments for cell signaling events. PDEδ (a.k.a. PDE6δ) is a prenyl-binding protein involved in intracellular localization of Ras-like membrane bound proteins within the cytosol9,10. PDEδ’s beta sandwich fold composes a hydrophobic pocket which binds to K-Ras farnesyl tails, allowing the complex to be solubilized in the cytosol for trafficking to the acceptor membrane6-10. K-Ras is released at the acceptor membrane upon binding of GTP-bound Arl2/3 to PDEδ6-9 (Fig. 1).
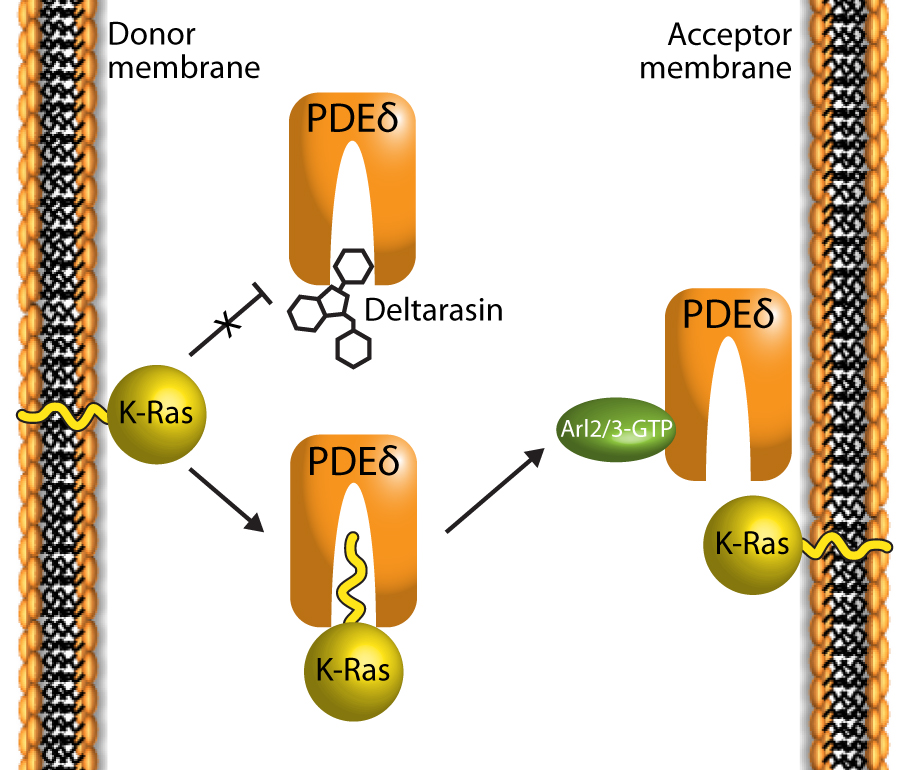
Figure 1: Deltarasin (a benzimidazole-based compound) inhibits the interaction of K-Ras and PDEδ by binding in the PDEδ pocket. K-Ras typically binds to PDEδ via the farnesylated lipid PTM at its C-terminus 5,6,8.
High-throughput screening has identified the benzimidazole-based compound Deltarasin as a disrupter of the interaction of PDEδ and K-Ras5. Deltarasin occupies the farnesyl-binding cavity, preventing the lipid tail of the Ras protein from binding to PDEδ; hence altering the subcellular localization of K-Ras. Cancer cells dependent upon K-Ras signaling for survival show increased cell death when treated with deltarasin. Furthermore, deltarasin treatment effectively reduces tumor growth rates in mice with tumor cell xenografts5. These findings support the importance of the Ras/PDEδ interaction on cancer development5. Interestingly, Ras protein function can remain intact without PDEδ11, possibly due to redundant cytosolic chaperones acting in the absence of PDEδ6,7.
Lipid Modification of Rho: A PTM’s Role in Cancer
Similar to the Ras GTPases, Rho GTPases (isoforms A, B, & C) are intimately involved in cancer cell morphogenesis and migration2,12,13. Furthermore, the PTMs prenylation and/or palmitoylation also regulate Rho GTPase trafficking and activity2. Like Ras, Rho proteins can be modified by covalent addition of a farnesyl or geranylgeranyl lipid and/or a palmitoyl lipid at the CAAX tetrapeptide motif14. Such modifications allow trafficking of Rho proteins to membranes, a necessity for normal biological activity2. Given the importance of lipid-based PTMs in the Ras and Rho signaling pathways, their inhibition is considered a promising target for cancer treatments14. Roberts et al.14 demonstrated that the CAAX motif is involved in proper Rho localization and activity through the use of various pharmacological inhibitors of farnesyltransferase, Rce1, and Icmt. These results strongly suggest that the Rce1 and Icmt CAAX-processing enzymes are important targets for therapeutic cancer inhibitors14.
As researchers reveal additional Ras and Rho regulatory mechanisms, methods to measure activated levels of these proteins take on increasing importance. To aid researchers with identifying and characterizing cancer therapeutics, Cytoskeleton, Inc. offers many small G-protein tools such as pull-down and G-LISA® activation assays which are widely used to measure the activated levels of small G-proteins. Pull-down methods measure activated protein levels utilizing a domain of an effector protein coupled to agarose beads and the activated level of the protein is measured by Western blot. G-LISA activation assays are faster and more sensitive than traditional pull-down techniques. G-LISAs require much less cell material and provide numerical data which allow easy comparison between samples. Small G-protein antibodies, activators, and inhibitors are also available to help elucidate Rho and Ras activation pathways.
Rho and Ras Research Tools
| Protein | Source | Purity | Cat. # | Amount | ||||
H-Ras His Protein, wild-type | >80% | 1 x 100 µg | ||||||
RhoA His Protein, wild-type | >80% | 1 x 100 µg | ||||||
RhoC His Protein, wild-type | >90% | 1 x 100 µg | ||||||
| Rhotekin-RBD Protein | >90% | RT01-A RT01-B | 1 x 500 µg 3 x 500 µg | |||||
| Rhotekin-RBD Beads | >85% | 2 x 2 mg | ||||||
Raf-RBD Beads | >85% | RF02-A RF02-B | 1 x 2 mg 4 x 2 mg | |||||
| Kit | Cat. # | Amount | ||||||
Ras G-LISA® Activation Assay, colorimetric | BK131 | 96 assays | ||||||
RhoA G-LISA® Activation Assay, colorimetric | BK124 | 96 assays | ||||||
RhoA G-LISA® Activation Assay, luminescence | BK121 | 96 assays | ||||||
Total RhoA ELISA | BK150 | 96 assays | ||||||
RhoGAP Assay Biochem Kit™ | BK105 | 80-160 assays | ||||||
RhoGEF Exchange Assay Biochem Kit™ | BK100 | 60-300 assays | ||||||
| G-protein Modulator | Cat. # | Amount | ||||||
Rho Activator II, Cell Permeable | CN03-A CN03-B | 3 x 20 µg 9 x 20 µg | ||||||
Rho Inhibitor I, Cell Permeable | CT04-A CT04-B | 1 x 20 µg 5 x 20 µg | ||||||
References
- Castellano E. and Santos E. 2011. Functional specificity of Ras isoforms: So similar but so different. Genes Cancer. 2: 216-231.
- Ridley A.J. 2013. RhoA, RhoB and RhoC have different roles in cancer cell migration. J. Microsc. doi: 10.1111/jmi.12025.
- Schubbert S., et al. 2007. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer. 7: 295-308.
- Bos J.L. 1989. ras oncogenes in human cancer: A review. Cancer Res. 49: 4682-4689.
- Zimmermann G., et al. 2013. Small molecule inhibition of the KRAS-PDEδ interaction impairs oncogenic KRAS signalling. Nature. 497: 638-642.
- Philips M.R. 2012. Ras hitchhikes on PDE6δ. Nat. Cell Biol. 14: 128-129.
- Chandra A., et al. 2012. The GDI-like solubilizing factor PDEδ sustains the spatial organization and signalling of Ras family proteins. Nat. Cell Biol. 14: 148-158.
- Iwig J.S. and Kuriyan J. 2013. Fixing a hole where the Ras gets in. Cell. 153: 1191-1193.
- Hanzal-Bayer M., et al. 2002. The complex of Arl2-GTP and PDEδ: From structure to function. EMBO J. 21: 2095-2106.
- Nancy V., et al. 2002. The delta subunit of retinal rod cGMP phosphodiesterase regulates the membrane association of Ras and Rap GTPases. J. Biol. Chem. 277: 15076–15084.
- Zhang H., et al. 2007. Deletion of PrBP/δ impedes transport of GRK1 and PDE6 catalytic subunits to photoreceptor outer segments. Proc. Natl. Acad. Sci. U.S.A. 104: 8857-8862.
- Hakem A., et al. 2005. RhoC is dispensable for embryogenesis and tumor initiation but essential for metastasis. Genes Dev. 19: 1974-1979.
- Huang M. and Prendergast G.C. 2006. RhoB in cancer suppression. Histol. Histopathol. 21: 213-218.
- Roberts P.J., et al. 2008. Rho Family GTPase modification and dependence on CAAX motif-signaled posttranslational modification. J. Biol. Chem. 283: 25150-25163.