Utilization of Newly Developed Mouse Monoclonal Anti-Acetyl Lysine Antibodies in Immunofluorescence Applications
We have recently developed two mouse monoclonal anti-acetyl-lysine (Ac-K) antibodies that have demonstrated excellent ability in visualizing acetylated proteins. The antibodies were raised against a proprietary mixture of acetylated proteins designed to optimize acetyl-lysine recognition for a wide range of sequence motifs. Data presented here show that both 7B5A1 (AAC02) and 19C4B2.1 (AAC03) provide robust staining of acetylated proteins by IF, which was highly specific for Ac-K detection as Ac-BSA competition inhibited all staining by either antibody. AAC03 produced the conventional Ac-K detection pattern, while AAC02 detected acetylated mitochondrial proteins by IF, which is unique relative to other commercial reagents. AAC02 IF staining co-localized with Mitotracker, but not lysosomal marker (Lamp-1), and were responsive to stimulants that are known to alter the mitochondria state. In immunoblot experiment, AAC02 detected multiple acetylated proteins in mitochondrial extracts. To further validate these findings mitochondrial target proteins were enriched and detected using these new Ac-K detection tools. AAC02 provides researchers with a unique avenue for studying acetylation in the mitochondria, and both antibodies are essential IF tools for investigating protein acetylation.

Figure 1 Legend: Swiss 3T3 cells were treated with TSA (1 mM for 6 h) and stained as described in the methods. Acetylated proteins were visualized using a green fluorescent secondary. Actin fibers were visualized using a red Rhodamine Phalloidin and the nucleus was stained with DAPI. The acetylated microtubule network is clearly visible with TSA-treatment, while the green fluorescent nuclear intensity indicate the high abundance of acetylated proteins in the nucleus. Acetylated BSA (10mg/ml) was used to compete for AAC02 binding as an indicator of AAC02 specificity for acetyl-lysine modifications.
Introduction
Lysine acetylation is a critical post-translational modification that has been shown to play a fundamental role in epigenetic histone regulation1. Recent acetylome studies indicate that acetyl-lysine regulation expands well beyond histones, and can target thousands of proteins and many different cellular processes. Many of these acetylome studies often link changes in global acetylation to disease progression2. Of interest, changes in acetylated microtubule and mitochondrial protein profiles are linked to many disease pathologies3,4. In particular, hyperacetylation of mitochondrial proteins appear to be a critical marker of metabolic disease5. Thus, having tools to quickly access the acetylated mitochondrial state may be beneficial for more effective investigation, diagnosis, and/or treatment of these diseases. Here, we present immunofluorescence (IF) validation data for two newly developed Ac-K antibodies AAC02 and AAC03, and their usefulness in visualizing acetylated proteins in general with a bias for mitochondrial and microtubule proteins.
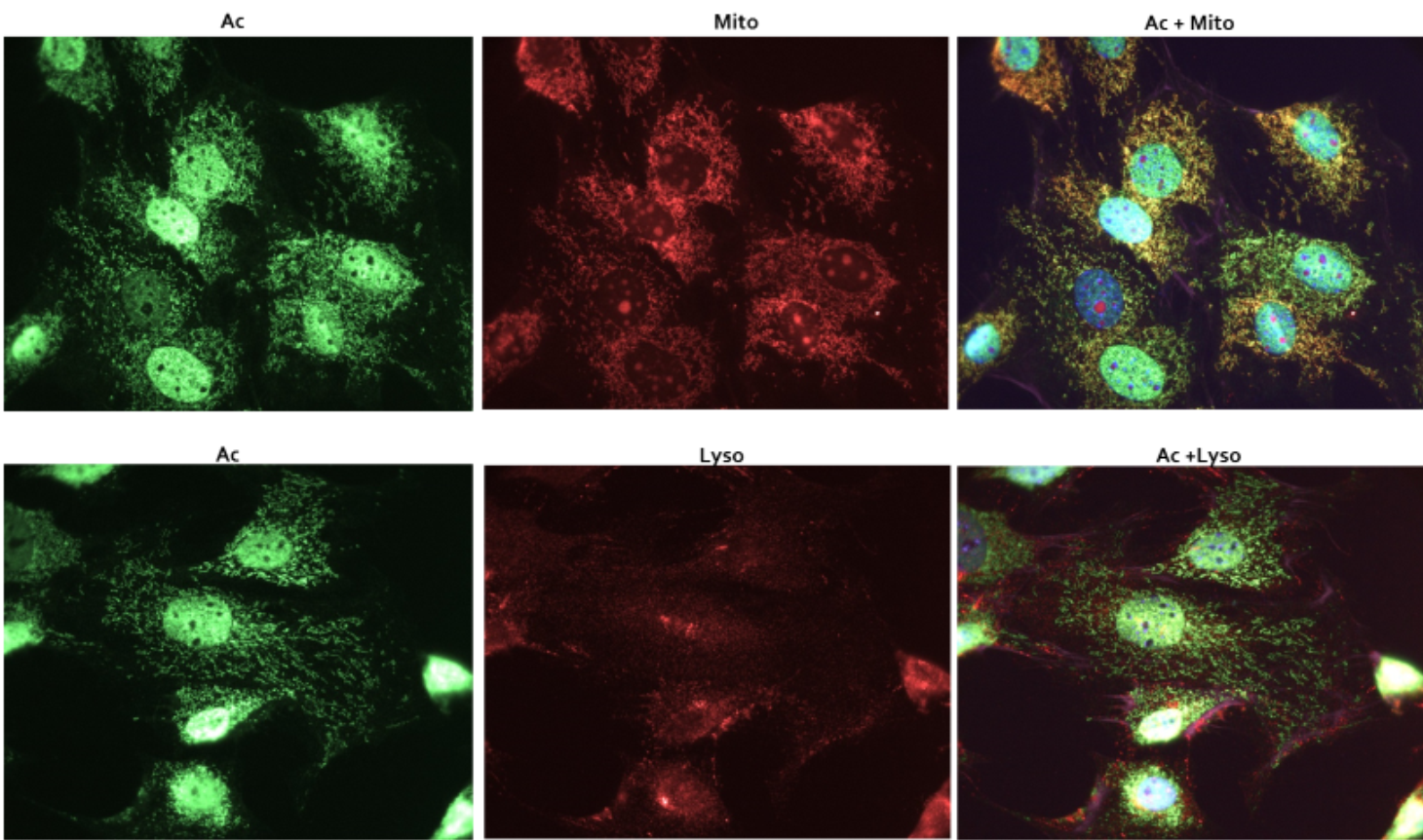
Figure 2 Legend: Swiss 3T3 cells were stained as described in the methods. Acetylated proteins were visualized in green fluorescence. Mitochondria were visualized with MitoTracker orange (Thermo Fisher), while the lysosome were visualized with anti-Lamp1 antibody (Abcam) Merged images of mitochondrial and acetylation signals, or lysosomal and acetylation signals were performed to define vesicular staining by AAC02
Results
AAC02 and AAC03 Ac-K antibodies, were recently validated for a variety of applications such as western blot and immunoprecipitation, and was shown to out perform other commercial reagents (see anti-acetyl-lysine antibodies white paper). Here, the ability of these antibodies to effectively bind acetylated proteins in an IF format was investigated. Figure 1 shows the detection of acetylated proteins by IF from 3T3 cells treated with 1mM TSA.
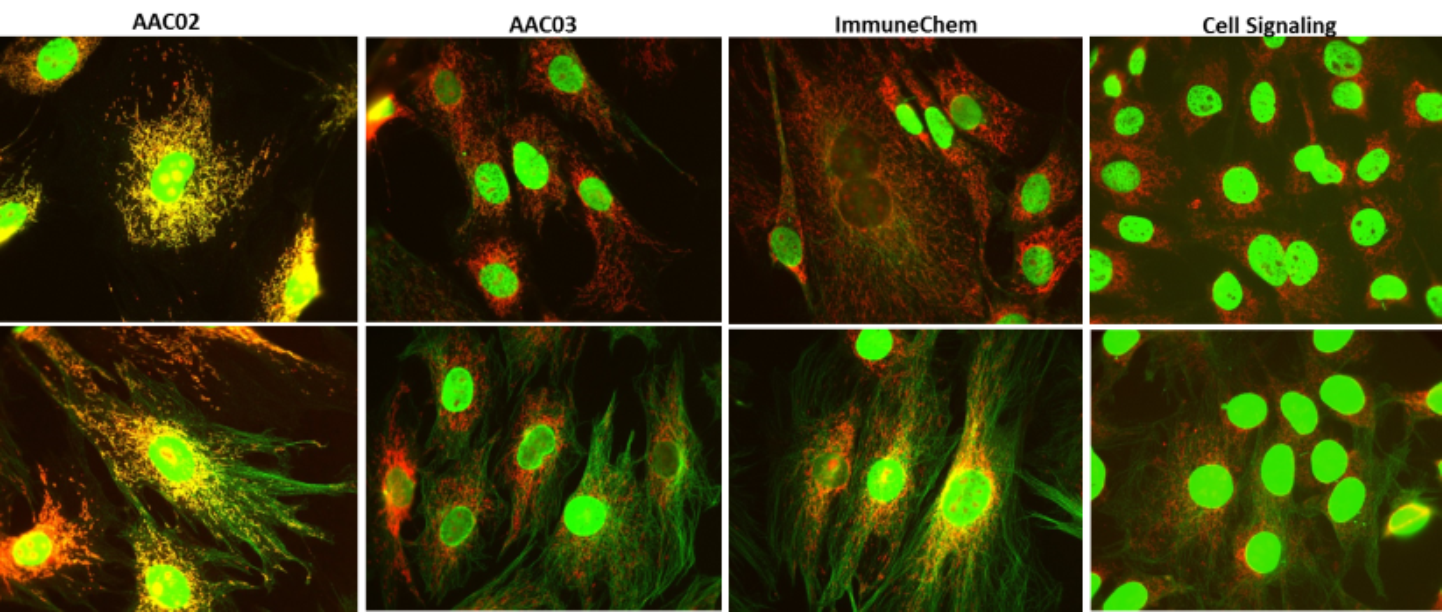
Figure 3 Legend: Untreated (top panel) and TSA treated (bottom panel) Swiss 3T3 cells were stained as described in the methods. Acetylated proteins were detected with AAC02, AAC03 and two other commercial available anti-Ac-K antibodies from ImmuneChem and Cell Signaling. Acetylation signals were visualized in green fluorescence. Mitochondria were visualized with MitoTracker orange (Thermo Fisher). Figure 3 showed merged images of mitochondrial and acetylation signals and the uniqueness of AAC02 in detecting mitochondrial acetylation compared to other Ac-K antibodies.
Both anti-Ac-K antibodies detect acetylated histones in response to TSA; however, AAC03 had robust acetylated microtubule detection, while AAC02 had weaker microtubule detection with distinct punctate vesicular like staining (Figure 1). To determine if AAC02 and AAC03 specifically bound Ac-K, the IF was performed in the presence of acetylated-BSA. The addition of high levels of Ac-BSA was intended to compete for anti-Ac-K antibody binding. Figure 1 provides evidence that in the presence of acetylated-BSA no Ac-K staining occurs, which suggests that AAC02 and AAC03 specifically binds to Ac-K.

Figure 4 Legend: A431 treated with 100uM H2O2 at different time intervals(0, 2, 6, and 24 hours) were stained as described in the methods. Acetylated proteins were visualized in green fluorescence. Mitochondria were visualized in red fluorescence using mitochondria marker (anti-Lamp1, Abcam). Merged images identified co-localization of mitochondrial and acetylation signals. Our results showed that AAC02 detected dynamics changes of mitochondrial acetylation in A431 cells with H2O2 treatment.
To further determine which acetylated vesicular structures were being detected by AAC02, IF experiments were performed with Mitotracker orange as well as a lysosomal marker (Lamp-1 antibody). Figure 2 shows robust co-localization between AAC02 staining and Mitotracker orange marker, but minimal co-localization with the lysosomal marker. These data suggests that the vesicles being stained by AAC02 may be mitochondria. These data are interesting as acetylome studies identify the mitochondria an organelle containing a large number of acetylated proteins.
Next, AAC02 and AAC03 were tested against other commercial anti-Ac-K reagents from Cell Signaling and ImmuneChem to compare their IF detection abilities. Results shown in figure 3, using the various antibodies showed that only AAC02 detected acetylated mitochondrial organelles by IF. These data further highlight the unique feature of AAC02 to visualize acetylated mitochondrial proteins by IF. Of note, AAC03 showed robust acetylation detection of microtubules compared to other commercial anti-Ac-K reagents (figure 3).
Mitochondrial protein acetylation has been shown to be highly responsive to metabolic stimulants, and other agents that regulate metabolic and/or mitochondrial function. To test the ability of AAC02 to track these dynamic and physiologic changes, cells were treated with H2O2. Figure 4 shows that AAC02 detects a decrease in acetylated mitochondrial proteins by IF, which is evident by the loss of co-localization between Ac-K signal and the mitochondria marker hexokinase. These changes are dynamic as the loss of Ac-K signal in the mitochondria occurs by 2 hrs and appears to recover, at least partially, by 24 hrs.
Mitochondrial fractionation studies were performed on A431 cells treated with H202 to determine if the changes observed by IF track with conventional methods used to study total mitochondrial acetylated proteins. The results in figure 5 shows acetylation profiles of cytoplasmic, nuclear, and mitochondrial proteins that have been treated with H202. The decrease in total mitochondrial protein acetylation, in response to H202, observed by western analysis is modest, but provides correlative evidence that H202 decreases the total acetylated protein profile in the mitochondria.
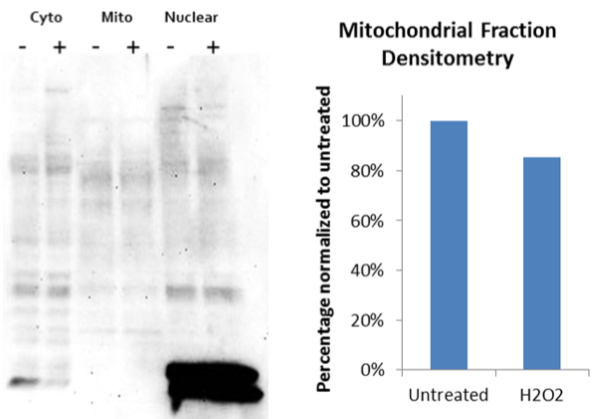
Figure 5 Legend: A431 cells were either untreated (-) or treated (+) with 100uM H2O2 for 2 hours. Mitochondria were isolated and solubilized as described in the methods. Sample extracts from different cellular compartments were resolved in SDS-PAGE gel and then transferred to a PVDF membrane. Acetylated proteins were detected with AAC02.
A final validation of AAC02 to detect mitochondrial proteins were performed by enriching for mitochondrial acetylated proteins using Cytoskeleton’s AAC04 enrichment beads. These beads were developed by crosslinking AAC02 and AAC03 to protein G agarose beads. A431 cells, untreated or treated with H2O2 were lysed with BlastR buffer. 1mg of total lysate were combined with AAC04 affinity beads, which resulted in acetylated protein enrichment. Samples were then probed with the following mitochondrial proteins: pyruvate dehydrogenase (PDH1), trifunctional protein alpha subunit (HADHA), long chain acyl-CoA dehydrogenase (ACADL), and glutamate dehydrogenase (GDH1), all of which has been shown to be acetylated. The data in figure 6 shows that the AAC04 affinity beads significantly enriched for acetylated mitochondrial proteins. These data support the IF results showing a decrease in mitochondrial protein acetylation in response to H2O2; furthermore, they provide the ability to detect which mitochondrial proteins are responding to the treatment.

Figure 6 Legend: Lysates from A431 cells either treated (+) or untreated (-) with H2O2 for 2 hours were obtained using BlastR buffer and filter system. Immunoprecipitation of acetylated proteins from 1 mg of lysates with AAC04 beads were performed as described in the methods. Eluted proteins were resolved in a SDS-PAGE gel and then transferred to a PVDF membrane. Change in acetylation level of mitochondrial proteins were examined with antibodies against specific mitochondrial proteins. 10 ug of total lysates were as input.
Summary
Two high-affinity pan-Ac-K antibodies, AAC02 and AAC03, were developed and compared to established Ac-K reagents. These data show that these highly optimized reagents perform as well as other for detecting acetylated proteins, and are superior for enrichment of endogenous, acetylated proteins in cell and tissue lysates. Importantly, AAC02 shows a unique ability in detecting mitochondrial acetylation by IF, providing a simple and quick assessment of mitochondrial acetylation status in cells. Importantly, the AAC04 beads allow for the enrichment of a representative pool of acetylated proteins without using the time consuming and labor intensive subcellular fractionation. Thus, providing an Ac-K toolset for identifying post-translational modification of mitochondrial proteins by acetylation.
Materials and Methods
Cell Culture and Reagents
Swiss 3T3 and A431 cells were grown in DMEM media (ATCC, VA) supplemented with 10% FBS (Altas Biologials, CO) and penicillin/streptomycin (ThermoFisher, MA). Trypsin/EDTA was obtained from Gibco (ThermoFisher, MA). BlastR lysis buffer and filter system (Cytoskeleton) was used to obtain a complete whole cell lysates for enrichment of acetylated proteins. Anti-acetyl lysine rabbit polyclonal antibody was from Cell Signaling. Anti-acetyl lysine rabbit polyclonal antibodies and bead conjugates were from ImmuneChem. Goat anti-mouse Alexa 488 and goat anti-rabbit Alexa 555 were from Invitrogen. Mitotracker orange was from ThermoFisher. Hydrogen peroxide and nicotinamide were from Sigma. TSA was from Cayman Chemical. Anti-lamp1 and anti-hexokinase 1 antibodies were from Abcam.
Mitochondria isolation from A431 cells
A431 cells either treated or untreated with H2O2 were collected in PBS and then resuspended in mitochondria isolation buffer (210mM mannitol, 70mM sucrose, 5mM Tris-HCl pH 7.5, 1mM EDTA) supplemented with proteases inhibitor cocktail (Cytoskeleton, CO). Cell suspension was transferred to a glass-Teflon potter and homogenized (5-10 stokes) at 1,600 rpm. The homogenate was transferred to a clean microtube and centrifuged at 3,000 rpm for 12 minutes. Supernatant was transferred to a clean microtube and centrifuged at 9,500 rpm for 10 minutes. Supernatant (cytostol fraction) was transferred to a new microtube and the pellet containing the mitochondria was washed 2 times with mitochondria isolation buffer. Mitochondria pellet can be stored at –80oC until use.
Immunoblot analysis of Mitochondrial Extract
Purified mitochondria from A431 cells either treated or untreated with H2O2 were lysed with ice-cold BlastR lysis buffer (Cytoskeleton, CO) containing a cocktail of TSA, nicotinamide and protease inhibitors (PIC02) (Cytoskeleton, CO). After dilution with BlastR dilution buffer, protein concentrations were determined with protein reagent, ADV02 (Cytoskeleton, CO), and measured at 600nm OD. Protein lysate samples were separated using Tris-glycine SDS-polyacrylamide gel electrophoresis (Thermo Fisher, MA) and transferred to Immobilon- P membranes (Millipore, MA). Membranes were blocked for 1 hr at room temperature in Tris-buffered saline (10 mM Tris-HCl, pH 8.0, 150 mM NaCl) containing 0.05% Tween-20 (TTBS) and 3% milk (Thrive Life, UT), and then incubated with 3% milk in TTBS solution containing primary antibodies for 1 hrs at room temperature (RT). Membranes were washed in TTBS 3x10minutes, prior to secondary antibody for 1hr at RT. Bound antibodies were visualized with horseradish peroxidase-coupled secondary antibodies and chemiluminescent reagent (Cytoskeleton, CO) according to the manufacturer’s directions.
Immunofluorescence Analysis of Mitochondrial Acetylation
Cells were seeded on glass coverslips for at least 24 hours before treatment. For Swsis 3T3 experiment, cells were either treated or untreated with 1uM of TSA for 6 hours. Mitotracker orange (100nM) was added to cells and incubated for 30 minutes before fixation and the rest of steps as shown below. For competition experiment, 10ug/ml acetylated BSA was added to AAC02 and AAC03 to compete for acetylation signals. For A431 experiment, cells were either treated or untreated with 100uM of H2O2 for 2 hours. Cells were then fixed with 4% formaldehyde for 10 minutes followed by permeabilization with 0.5% triton x-100 for 15 minutes. Cells were blocked with 2% BSA/PBST/5% normal goat serum for 30 minutes. Primary antibodies against acetylated proteins and mitochondrial hexokinase 1 were used at 1:500 and 1:100 respectively in 2% BSA/PBST. Acetylation signals and mitochondrial signals were visualized with goat anti-mouse Alexa 488 and goat anti-rabbit Alexa 555 respectively.
Immunoprecipitation Assay
A431 cells, treated or untreated with 100uM H2O2, were lysed with ice-cold BlastR lysis buffer containing a cocktail TSA(1uM), nicotinamide(16.5mM), and protease inhibitors (PIC02). DNA was removed by passing the lysate through the BlastR filter system (Cytoskeleton, CO). After dilution with BlastR dilution buffer, protein concentrations were determined with ADV02 and measured at 600nm OD. 50ul of AAC04 beads were added to 1 mg of lysate for 1-2 hr at 4°C on an end-over- end tumbler. After incubation, the affinity beads from each sample were pelleted, and washed 3X with BlastR wash buffer. Bound proteins were eluted using bead elution buffer (Cytoskeleton, CO) and detected by immunoblotting.
References
- Verdin E, Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat Rev Mol Cell Biol. 2015;16(4):258-64.
- Iyer A, Fairlie DP, Brown L. Lysine acetylation in obe sity, diabetes and metabolic disease. Immunol Cell Biol. 2012;90(1):39-46.
- Li L, Yang XJ. Tubulin acetylation: responsible enzymes, biological functions and human diseases. Cell Mol Life Sci. 2015;72(22):4237-55.
- Wagner GR, Payne RM. Mitochondrial acetylation and diseases of aging. J Aging Res. 2011;2011:234875.
- Horton JL, Martin OJ, Lai L, et al. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight. 2016; 2(1):1-14.
Product Citations/Related Products
Acetyl-Lysine Antibody Mouse Monoclonal (7B5A1) (Cat. # AAC02)
Acetyl-Lysine Antibody Mouse Monoclonal (19C4B2.1) (Cat. # AAC03)