Utilization of newly developed mouse monoclonal anti-acetyl lysine antibodies in western blot and immunoprecipitation applications
We have recently developed two mouse monoclonal anti-acetyl lysine (Ac-K) antibodies that have demonstrated excellent ability in enriching acetylated proteins from cell and tissue extracts. The antibodies were raised against a proprietary mixture of acetylated proteins designed to optimize acetyl lysine recognition for a wide range of sequence motifs. Our validation data showed that both 7B5A1 and 19C4B2.1 immunoprecipitated a broad range of acetylated proteins, with unique specificity, from cells treated with trichostatin A (TSA) and nicotinamide. A more complete enrichment profile was observed when both clones were used together in immunoprecipitation validation experiments. For western blot applications, clone 7B5A1 detected more acetylated proteins than clone 19C4B2.1. Both clones showed an enhanced ability to detect acetylated proteins when conjugated to horseradish peroxidase (HRP). Compared to other commercial Ac-K reagents, clones 7B5A1 and 19C4B2.1 were superior at immunoprecipitating acetylated proteins. They also detected a slightly different acetylated protein profile in western blot analysis. Currently, both clones have also been validated for immunofluorescence staining applications (see separate white paper for this data).


Introduction
Acetylation of proteins can occur as a co-translational or post-translational modification (PTM) (1). Co-translational acetylation occurs at the N-terminus of approximately 85% of mammalian proteins, is irreversible, and is thought to be important in protein stability, localization, and synthesis (1). Post-translational acetylation occurs on the epsilon amino group of lysine residues as a reversible and highly dynamic PTM that is known to be a key regulator in multiple cellular events, including chromatin structure, transcription, metabolism , signal transduction, and cytoskeletal regulation (2-3). Identification and enrichment of acetylated proteins have been hindered by a lack of high affinity Ac-K antibodies. Here, we present validation data for two newly developed Ac-K antibodies, 7B5A1 and 19C4B2.1, and their usefulness in enriching and detecting acetylated proteins in both cell and tissue lysates.

Figure 1a Legend: Cos cells were untreated (-) or treated (+) with 1uM TSA and 1mM nicotinamide for 6 hours. Whole cell lysates were obtained using BlastR lysis buffer and filter system (Cytoskeleton). 30ug of lysates were resolved by SDS-PAGE and proteins were transferred to PVDF membrane and analyzed with western blot by 7B5A1-HRP(1:4000), 19C4B2.1-HRP (1:4000), and AAC01-HRP (1:4000), respectively.
Figure 1b Legend: Ac-K antibodies conjugated beads (40ug) were used to immunoprecipitate acetylated proteins from Cos cell lysates as shown above. Captured proteins were eluted and resolved by SDS-PAGE and then transferred to PVDF membrane. 19C4B2.1-HRP (1:2000) was used to detect enriched acetylated proteins.
Results and Discussion
Two newly developed Ac-K antibodies were compared to Cat. # AAC01 (Cytoskeleton), an anti-acetyl lysine antibody, that has been validated for a variety of applications such as western blot, immunoprecipitation (IP), immunofluorescence staining and ChIP. Figure 1a shows the detection of acetylated proteins by western blot from Cos cells treated with 1mM TSA and 1mM nicotinamide.
For IP experiments, 7B5A1 and 19C4B2.1 were separately crosslinked to protein G beads (here after termed affinity beads) to minimize the co-elution of IP antibodies (heavy and light chain) during the final protein elution step. Interestingly, IP with 7B5A1 and 19C4B2.1 affinity beads from Cos cell lysates treated with TSA and nicotinamide resulted in slightly different acetylated protein isolation patterns (Figure 1b). Still, either antibody showed a more robust and higher enrichment of acetylated proteins relative to AAC01 affinity bead acetyl lysine enrichment. These data suggest that both 7B5A1 and 19C4B2.1 performed better than AAC01 in detecting and enriching acetylated proteins.
Next, 7B5A1 and 19C4B2.1 were tested against other commercial anti-acetyl lysine reagents from Cell Signaling and ImmuneChem to compare their detection abilities. Western blot analysis, using the various antibodies showed that both 7B5A1 and the rabbit antibody from cell signaling detected the most robust acetylation profile; however, the profiles differed slightly different. Of note, Both 7B5A1 and 19C4B2.1 showed enhanced detection signals when they were conjugated to HRP directly (Figure 2).
Both 7B5A1 and 19C4B2.1 showed slightly different enrichment patterns of acetylated proteins; thus, when combined the mixture of these affinity beads isolated a more complete acetylation profile (figure 3). We then took these beads and performed side-by-side IP comparisons with different Ac-K reagents. 1mg of cell lysate from Cos cells treated or untreated with TSA and nicotinamide were used per IP reaction. 7B5A1-19C4B2.1 bead mixture was used at 20 and 40ug per IP reaction. Ac-K affinity bead from ImmuneChem was used at 40 and 80ug. Ac-K rabbit monoclonal mix from Cell Signaling was used at 1:100 per manufacturer’s instruction. We didn’t include the Cell Signaling Ac-K mouse monoclonal antibody in IP experiment because it is not recommended for IP applications. 60ug of normal mouse IgG bead was included as a non-specific proteins binding control. The results suggested that our antibodies bead mixture performed exceptionally well in enriching a wide range of acetylated proteins whereas the other commercial Ac-K reagents could only enrich the most abundance acetylated proteins (e.g. acetylated tubulin and histones) (Figure 3).
Isolation of a robust and comprehensive acetylation profile is critical, but having the ability to identify acetylated target proteins is paramount for endogenous studies, validation studies, and for new target protein identification. Figure 4a demonstrates the detection of changes in the acetylation profile of four different proteins (EGFR, Hsp90, P53 and RhoGDI) from the enriched acetylated protein pool from A431 cells treated with TSA and nicotinamide.
Utilization of these antibodies and affinity beads are not limited to cell culture studies. Data provided here show IP of acetylated proteins from mouse liver and heart tissue that was lysed with BlastR lysis buffer (Cytoskeleton). Importantly, these affinity beads captured robust and unique acetylation profiles. The acetylated proteins were identified using the 19C4B2.1-HRP acetyl lysine antibody (Figure 4b).

Figure 2 Legend: 30ug of lysates from Cos cells untreated (-) or treated (+) with 1uM TSA and 1 mM nicotinamide were resolved by SDS-PAGE and proteins were transferred to PVDF membrane. To detect acetylated proteins, blots were probed with (1) 7B5A1 (1:500), (2) 7B5A1-HRP (1:2000), (3) 19C4B2.1 (1:500), (4) 19C4B2.1-HRP (1:2000), (5) Ac-K mouse monoclonal (Cell Signaling, 1:1000), (6) Ac-K rabbit monoclonal mix (Cell Signaling, 1:1000), and (7) Ac-K rabbit polyclonal (ImmuneChem, 1:1000), respectively. (Red circle) Acetylated tubulin. (Red triangle) Acetylated histones.
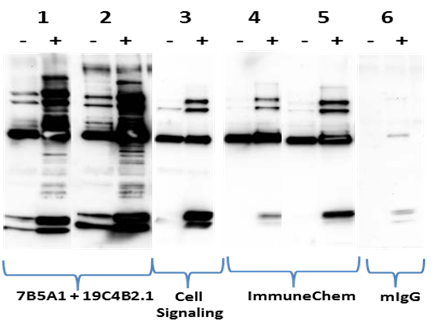
Figure 3 Legend: 1mg of lysates from Cos cells either untreated (-) or treated (+) with 1uM TSA and 1mM nicotinamide were used in IP reactions with different Ac-K reagents as indicated: (1) 7B5A1-19C4B2.1 bead mixture (20ug) (2) 7B5A1-19C4B2.1 bead mixture (60ug) (3) Ac-K rabbit monoclonal mix (Cell Signaling, 1:100) (4) Ac-K bead (ImmuneChem, 40ug) (5) Ac-K bead (ImmuneChem, 80ug) (6) mouse IgG bead (60ug). Captured proteins were eluted and resolved by SDS-PAGE and analyzed by western blot with 19C4B2.1-HRP antibody (1:2000).

Figure 4a Legend: A431 cells, untreated (-) or treated (+) with 1uM TSA and 1mM nicotinamide for 6 hours, were isolated using BlastR buffer. IP was performed using 7B5A1-19C4B2.1 bead mixture (60ug). Total cell lysate (Input) and immunoprecipitated samples were separated by SDS-PAGE and analyzed by western blot with antibodies against EGFR (Millipore, 1:1000), Hsp90 (Abcam, 1:20,000), P53 (Sigma, 1:2000), and RhoDGI (Millipore,1:1000).
Figure 4b Legend: Mouse tissue extracts (liver and heart) were obtained with BlastR buffer. IP was performed using 7B5A1-19C4B2.1 bead mixture (60ug) or mIgG control bead (60ug) in 1mg of tissue extracts. Enriched proteins were separated by SDS-PAGE and analyzed by western blot with 19C4B2.1-HRP (1:2000).
Summary
Two high-affinity pan-Ac-K antibodies, clone 7B5A1 and 19C4B2.1, were developed and compared to established Ac-K reagents. These data show that these highly optimized reagents perform as well as other for detecting acetylated proteins, and are superior for enrichment of endogenous, acetylated proteins in cell and tissue
lysates. Importantly, the enrichment bead formulation was optimized to minimize heavy and light chain interference, while simultaneously providing the most robust acetylated protein profile. This was achieved in part by accounting for the existence of high-abundance acetylated histones that can often saturate other Ac-K enrichment tools. In doing so, this allowed for the enrichment of a representative pool of acetylated proteins without using subcellular fractionation. Of importance, these data demonstrated that detection of physiologic changes in acetylation of multiple target proteins was possible using the 7B5A1-19C4B2.1 bead mixture. The enrichment, isolation, and detection of these acetylated target proteins occurred in the same enriched protein pool, instead of conducting IP for each protein as would need to be done using protein specific antibody enrichment. This level of enrichment speaks to the quality of the IP capabilities that these antibodies posses. In conclusion, 7B5A1 and 19C4B2.1 antibodies performed exceptionally well compared to other commercial Ac-K reagents; especially, in IP applications. We believe that our Ac-K antibodies and enrichment affinity beads are a great discovery tool for identifying post-translational modification of proteins by acetylation.
Materials and Methods
Cell Culture and Reagents
Cos and A431 cells were grown in DMEM media (ATCC, VA) supplemented with 10% FBS (Altas Biologials, CO) and penicillin/streptomycin (ThermoFisher, MA). Trypsin/EDTA was obtained from Gibco (ThermoFisher, MA). For treatment, cells (either Cos or A431) were grown to about 70% confluent and then treated with 1uM TSA (Cayman Chemical, MI) and 1mM nicotinamide (Sigma, MO) for 6 hours. BlastR lysis buffer and filter system (Cytoskeleton) was used to obtain a complete whole cell lysates for western blot analysis and enrichment of acetylated proteins. Anti-acetyl lysine mouse monoclonal antibody and rabbit monoclonal mix antibodies were from Cell Signaling. Anti-acetyl lysine rabbit polyclonal antibodies and bead conjugates were from ImmuneChem. Antibodies to EGFR and RhoGDI were from Millipore. Antibody to P53 was from Sigma. Antibody to Hsp90 was from Abcam.
Western blotting
Cos cells were lysed with ice-cold BlastR lysis buffer (Cytoskeleton, CO) containing a cocktail of TSA (1uM), nicotinamide (16.5mM) and protease inhibitors (PIC02) (Cytoskeleton, CO). DNA was removed by passing the lysate through the BlastR filter system (Cytoskeleton, CO). After dilution with BlastR dilution buffer, protein concentrations were determined with protein reagent, ADV02 (Cytoskeleton, CO), and measured at 600nm OD. Protein lysate samples were separated using Tris-glycine SDS-polyacrylamide gel electrophoresis (ThermoFisher, MA) and transferred to Immobilon- P membranes (Millipore, MA). Membranes were blocked for 1 hr at room temperature in Tris-buffered saline (10 mM Tris-HCl, pH 8.0, 150 mM NaCl) containing 0.05% Tween-20 (TTBS) and 5% milk (Thrive Life, UT), and then incubated with 3% milk in TTBS solution containing primary antibodies for 1 hrs at room temperature (RT). Membranes were washed in TTBS 3x10minutes, prior to secondary antibody for 1hr at RT. Bound antibodies were visualized with horseradish peroxidase-coupled secondary antibodies and chemiluminescent reagent (Cytoskeleton, CO) according to the manufacturer’s directions.
Immunoprecipitation assay
Cos and A431 cells were lysed with ice-cold BlastR lysis buffer containing a cocktail TSA (1uM), nicotinamide (16.5mM), and protease inhibitors (PIC02). DNA was removed by passing the lysate through the BlastR filter system (Cytoskeleton, CO). After dilution with BlastR dilution buffer, protein concentrations were determined with ADV02 and measured at 600nm OD. For tissue lysate preparation, mouse liver or heart was lysed at 1 mL of BlastR lysis buffer per 100mg of tissue. For these experiments 100mg of tissue was processed. After addition of 1mL of BlastR lysis buffer to 100mg of fresh tissue., the sample was placed into a homogenizer and 10-12 strokes were applied. The viscous tissue lysate was first passed through the BlastR filter to remove genomic DNA. Equal volume of BlastR dilution buffer was then added to the filtered lysate and gently mixed. Lysate was then spun in a microcentrifuge at 14k rpm for 15min at 4°C to pellet any remaining tissue debris. Additional BlastR dilution buffer was added to the recovered supernatant for a final dilution of 1:5. Protein concentrations were determined with ADV02 and measured at 600nm OD.
The appropriate amount of Ac-K reagents or mIgG control beads were added to 1 mg of lysate for 1-2 hr at 4°C on an end-over- end tumbler. After incubation, the affinity beads from each sample were pelleted, and washed 3X with BlastR wash buffer. Bound proteins were eluted using bead elution buffer (Cytoskeleton, CO) and detected by western immunoblotting.
References
- Bogdan P. and Sheman F. 2002. The diversity if acetylated proteins. Genome Biol. 3(5):reviews 0006.
- Lundby A. et al. 2012. Protemmicanalysis of lysine acetylation sites in rat tissues reveals organ specificity and cellular patterns. Cell Reports 2:419-431.
- Sadoul K. et al. 2010. The tale of protein lysine acetylation in the cytoplasm. J. Biomed. Biotech. 2011:1-15.
Related Products
New Signal-Seeker™ Acetyl-Lysine Detection Kit (30 assay) (Cat. # BK163)
New Signal-Seeker™ Acetyl-Lysine Detection Kit (10 assay) (Cat. # BK163-S)
New Acetyl-Lysine-HRP Antibody Mouse Monoclonal (19C4B2.1) (Cat. # AAC03-HRP)
New Acetyl-Lysine Control Beads (Cat. # CIG02-beads)
New Acetyl-Lysine Antibody Mouse Monoclonal (7B5A1) (Cat. # AAC02)
New Acetyl-Lysine Antibody Mouse Monoclonal (19C4B2.1) (Cat. # AAC03)
New Acetyl-Lysine Affinity Beads (Cat. # AAC04-beads)
Chemically Acetylated Bovine Serum Albumin (BSA) (Cat. # AACX1)