
Important Notice
This technical guide is an accompaniment to the G-LISA kit Protocol and is designed to provide additional information that may be of use when designing or troubleshooting an activation assay.
Click Here for PDF Version
Table of Contents
Section I:
Section II:
- Preparation of Lysates for G-LISA Assays
- Recommended lysate amount per assay
- Lysis buffer compatibility between assays
- Growth and treatment of tissue culture cell lines
- Preparation of lysates from 2D tissue culture cell lines
- Preparation of lysates from 3D tissue culture cell lines
- Preparation of lysates from tissue samples
- Equalization of cell lysate concentrations
Section V:
Section I: Introduction
Technical Guides are an accompaniment to the G-LISA kit Protocols and are designed to provide additional information to the end user that may be of use when designing or troubleshooting an activation assay.
Go online to cytoskeleton.supremeclients.com for the latest Technical Guide updates.
For assistance, contact Technical Support at 303-322-2254 or e-mail [email protected].
Section II: Preparation of Lysates for G-LISA
Recommended lysate amounts per assay
The G-LISA range of activation assays generally requires 6.25-50 µg of lysate per assay. Table 1 summarizes the recommended concentrations of lysate for each small G-protein (GTPase) target. The values in Table 1 are recommended starting points for optimizing a given G-LISA assay. Lysates that are too far above the recommended range may contain a high level of GAPs (GTPase activation proteins) that can inactivate the target GTPase, even in lysis buffer. Final lysate concentrations will vary depending upon experimental factors such as:
A) Total amount of a given GTPase in your cell line or tissue: In general cell lines or tissue that have a high endogenous level of the target GTPase will give a more robust response to an activator.
B) Degree of activation achieved under your experimental conditions: As a general guideline roughly 2-10% of total cellular small G-protein is activated in response to a given stimuli.
Table 1: Recommended lysate amounts for G-LISA assays
| Small G-protein target | Recommended lysate amount per G-LISA assay (µg) | Recommended lysate concentration range (mg/ml) | Volume of lysate per assay (µl) |
|---|---|---|---|
| Arf1 | 12.50 - 25.00 | 0.50 - 1.00 | 25 |
| Arf6 | 12.50 - 25.00 | 0.50 - 1.00 | 25 |
| Cdc42 | 7.50 - 50.00 | 0.15 - 1.00 | 50 |
| RalA | 6.25 - 25.00 | 0.25 - 1.00 | 25 |
| Rac1 | 12.50 - 50.00 | 0.25 - 1.00 | 50 |
| Rac1,2,3 | 6.25 - 25.00 | 0.25 - 1.00 | 25 |
| Ras (pan) | 6.25 - 25.00 | 0.25 - 1.00 | 25 |
| RhoA | 10.00 - 50.00 | 0.40 - 2.00 | 25 |
Lysis buffer compatibility between assays
Lysis buffer composition is critical to optimal performance of a G-LISA assay. In most assays a standard lysis buffer composition (GL36) has been used (see Table 2). This allows the end user to test various GTPase activities in the same lysate.
For Arf6 analysis one can dilute a GL36 lysate 1:1 with water to achieve the correct assay lysis buffer.
It should be noted that the Cdc42 lysis buffer (GL35) is not compatible with GL36 lysates.
Table 2: List of G-LISA Lysis buffers
| Small G-protein Target | Lysis Buffer in Kit | Notes |
|---|---|---|
| Arf1 | GL36 | Standard G-LISA buffer, compatible with most G-LISA assays. Composed of a proprietary formulation of Tris pH 7.5, MgCl2, NaCl, IGEPAL and SDS. |
| Arf6 | GL70 | Buffer composition is 0.5X GL36 |
| Cdc42 | GL35 | Identical components to GL36, composition varies as follows; Tris (1X GL36), MgCl2 (8X GL36), NaCl (2X GL36), IGEPAL (1X GL36), SDS (5X GL36) |
| RalA | GL36 | Standard G-LISA buffer, compatible with most G-LISA assays. Composed of a proprietary formulation of Tris pH 7.5, MgCl2, NaCl, IGEPAL and SDS. |
| Rac1 | GL36 | Standard G-LISA buffer, compatible with most G-LISA assays. Composed of a proprietary formulation of Tris pH 7.5, MgCl2, NaCl, IGEPAL and SDS. |
| Rac1,2,3 | GL36 | Standard G-LISA buffer, compatible with most G-LISA assays. Composed of a proprietary formulation of Tris pH 7.5, MgCl2, NaCl, IGEPAL and SDS. |
| Ras | GL36 | Standard G-LISA buffer, compatible with most G-LISA assays. Composed of a proprietary formulation of Tris pH 7.5, MgCl2, NaCl, IGEPAL and SDS. |
| RhoA | GL36 | Standard G-LISA buffer, compatible with most G-LISA assays. Composed of a proprietary formulation of Tris pH 7.5, MgCl2, NaCl, IGEPAL and SDS. |
Growth and treatment of tissue culture cells
The health and responsiveness of your cell line is the single most important parameter for the success and reproducibility of GTPase activation assays. Growth conditions vary widely between cell lines, therefore the reader is referred to Tables 3A-H for helpful references regarding GTPase activation in a variety of cell lines. Optimal GTPase activation/inhibition conditions in a given cell line will need to be determined empirically.
Some general guidelines are given below;
1) When possible, untreated samples should have low basal cellular levels of GTPase activity (controlled state). For example, serum starvation often results in low basal levels of GTPase activity and leads to a much greater response to a given activator (see Tables 3A-H).
2) GTPase activation is often very transient, lasting only seconds to minutes. A time courses of activation/inhibition and titration of activating or inhibiting factors are highly recommended when establishing assay conditions.
3) When possible cells should be checked for their responsiveness (responsive state) to a known stimulus (see Tables 3A-H).
4) Poor culturing technique such as sub-culturing of cells that have previously been allowed to become overgrown can result in essentially non-responsive cells. For example, Swiss 3T3 cells grown to >70% confluency should not be used for Rho GTPase activation studies.
5) Cell confluency can affect the cellular response of GTPases to activators/inhibitors. For example RhoA activation generally requires sub confluent cells for a robust response to activators. Confluency restrictions vary widely between different cell lines and different GTPases (see Tables 3A-H).
6) Once an inducible GTPase response has been achieved, it is particularly important to maintain consistent experimental conditions. An Experimental Record Sheet is included in this Guide (Data Analysis section) and is useful for tracking key experimental parameters.
Table 3A: Transfection-based GTP-Arf1 modulation
| Gene transfected | Treatment | Cell type used | Response | Assay Type | Ref. |
|---|---|---|---|---|---|
| Arf1-HA + GBF1 | 5-25 mg/ml BFA | Cos7 cells | GBF1 increased Arf1-GTP levels 2-3 fold and this activation was not inhibited by BFA. | GGA3 pull-down | 1 |
| HCV NS5A | N/A | Huh7 cells | Hepatitus C virus NS5A protein expression reduced Arf1-GTP levels | GGA3 pull-down | 2 |
| Ephrin-B1 | 2 mg/ml EphB2-Fc | Capan-1 cells Panc-1 cells | Treatment for 1.5 hr with EphB2-Fc resulted in elevated Arf1-GTP levels | GGA3 pull-down | 3 |
| Enterovirus 3A protein | N/A | Cos7 cells | Expression of enterovirus 3A protein decreased total Arf1-GTP levels by 60% | GGA3 pull-down | 4 |
Table 3B: Non-transfection-based GTP-Arf1 modulation
| Arf1 Modulator | Treatment | Cell type used | Response | Assay Type | Ref |
|---|---|---|---|---|---|
| Formyl methionyl-lysyl-phenylalanine (fMLF) | 5 ng/ml | Polymorphonuclear neutrophils (PMNs), PLB-985 cells | Treatment for 2 minutes with fMLF resulted in ~5-fold activation of Arf1 in PMNs. | GGA3 pull-down | 5 |
| QS11 (ARFGAP1 inhibitor) | 1-2.5 mM | NIH 3T3 cells | Treatment for 36 hr resulting in dose-dependent activation of Arf1 and Arf6 | GGA3 pull-down | 6 |
| Poliovirus | 10 PFU/cell | HeLa cells | Time-dependent increase in Arf1-GTP with a maximal activation of ~3.75-fold | GGA3 pull-down | 7 |
| Ephrin-B1 | 2 mg/ml EphB2-Fc | SUIT-4 cells | Treatment for 1.5 hr with EphB2-Fc resulted in elevated Arf1-GTP levels | GGA3 pull-down | 3 |
Table 3C: Modulators of GTP-Arf6 levels in cells
| Arf6 Modulator | Treatment | Cell type used | Response | Assay Type | Ref |
|---|---|---|---|---|---|
| Fibronectin (Fn) | 5 ng/ml Fn coated plates | Mouse embryonic fibroblasts (MEFs) | Control cells harvested and kept in suspension for 90 min. followed by replating to Fn-coated plates for 15-30 min. resulted in a 4-fold activation of Arf6 | GGA3 pull-down | 8 |
| Fibronectin (Fn) | 5 ng/ml Fn coated plates | NIH 3T3 cells | Control cells harvested and kept in suspension for 60 min. followed by replating to Fn-coated plates for 5 min. resulted in a modest activation of Arf6 | GGA3 pull-down | 9 |
| Formyl methionyl-lysyl-phenylalanine (fMLF) | 5 ng/ml | Polymorphonuclear neutrophils (PMNs), PLB-985 cells | Treatment for 2 min. with fMLF resulted in ~10-fold activation of Arf6 in PMNs, and a ~ 3.5-fold activation in PLB-985 cells | GGA3 pull-down | 5 |
| QS11 (ARFGAP1 inhibitor) | 1-2.5 mM | NIH 3T3 cells | Treatment for 36 hr resulting in dose-dependent activation of Arf6 | GGA3 pull-down | 6 |
| Collagen | 10 mg/ml | Platelets | Time-dependent decrease in Arf6-GTP levels with collagen treatment | GGA3 pull-down | 10 |
| Insulin | 100 nM | HepG2 cells | Treatment of cells for 20 min. with insulin resulted in ~3-fold increase in Arf6-GTP | GGA3 pull-down | 11 |
| HGF | 10 ng/ml | MDCK cells | Time-dependent increase in Arf6-GTP up to 16 hr. with ~30-fold maximal increase in Arf6-GTP | GGA3 pull-down | 12 |
| Semaphorin 3E | 100 ng/ml | SVEC cells | Time-dependent ~2-fold increase in Arf6-GTP | GGA3 pull-down | 13 |
Table 3D: Modulators of GTP-Cdc42 levels in cells
| Cdc42 Modulator | Treatment | Cell type used | Response | Assay Type | Ref |
|---|---|---|---|---|---|
| ML-7 | 20 µM | Primary rat microglia | Inhibition of Pyk2 mediated Cdc42 activation | Cdc42 G-LISA | 14 |
| Epidermal Growth Factor | 50 ng/ml | Mouse mesenchymal stem cells | 1.5 fold activation of Cdc42 | Cdc42 G-LISA | 15 |
| Ethanol | 30 mM | Rat hippocampal neurons | 35% reduction in Cdc42 activation | Cdc42 G-LISA | 16 |
Table 3E: Modulators of GTP-Rac levels in cells
| Rac Activator | Treatment | Cell Line Used | Response | Assay Type | Ref. |
|---|---|---|---|---|---|
| Epidermal Growth Factor | 50 ng/ml | US7MG human glioblastoma | 1.5 fold activation after 5 minutes with 2D cultures. 1.3 fold activation in 3D cultures | Rac G-LISA® | 17 |
| MCP-1 | 10 ng/ml | Murine alveolar macrophages | Maximal activation at 4h | Rac G-LISA® | 18 |
| Heregulin beta1 | 0-30 ng/ml | Breast cancer cell lines | Dose-dependent activation | PAK-PBD pulldown assay | 19 |
| Interleukin-3 | 5 µg/ml for 5 min | Primary bone marrow derived mast cells | 2 fold increase over control | PAK-PBD pulldown assay | 20 |
Table 3F: Modulators of GTP-RalA levels in cells
| RalA Modulator | Treatment | Cell type used | Response | Assay Type | Ref. |
|---|---|---|---|---|---|
| Epidermal growth factor | 50 ng/ml | Cos7 | FRET activity and lamellipodia induction | FRET | 21 |
Table 3G: Modulators of GTP-Ras levels in cell Ras Activator Treatment
| Ras Activator | Treatment | Cell line used | Response | Assay Type | Ref |
|---|---|---|---|---|---|
| Epidermal Growth Factor | 100 ng/ml 5 minutes | HeLa | Dose-dependent activation | Raf1-RBD pulldown assay | 22 |
| Hepatocyte Growth Factor | 100 ng/ml 5 minutes | HeLa | Dose-dependent activation | Raf1-RBD pulldown assay | 22 |
| IL-3 | 50 ng/ml 5 minutes | BaF3 | 14 fold activation after 5 minutes | Raf1-RBD pulldown assay | 24 |
| aCD3 + aCD28 | 5 mg/ml each 10 minutes | Jurkat | Dose-dependent activation | Raf1-RBD pulldown assay | 24 |
| PMA ionomycin | 100 ng/ml 500 ng/ml 10 minutes | Jurkat | Dose-dependent activation | Raf1-RBD pulldown assay | 24 |
Table 3H: Modulators of GTP-Rho levels in cell
| Rho Activator | Treatment | Cell line used | Response | Assay Type | Ref |
|---|---|---|---|---|---|
| Lysophosphatidylcholine (lysophospholipid) | 100 nM | Human melanocyte | After 1 min, 1.5 fold increase over control. Activation peaked at 5 min (5.3 fold increase) and declined to baseline by 60 min. | RhoA G-LISA (Cat.# BK124) | 25 |
| Calpeptin (Cat. # CN01) | 0.5 mU/ml | Uterine myocyte | After 15 min treatment, 2.4 fold increase over vehicle control | RhoA G-LISA (Cat.# BK124) | 26 |
| Bombesin | 10 nM | Swiss 3T3 cells | Maximal activation after 1 min which is sustained for at least 30 min | Actin morphology | 27 |
| Calpeptin | 100 µg/ml | REF-52 fibroblast & Swiss 3T3 cells | Maximal activation after 5 to 10 min with extended activation time up to 30 min, decreasing thereafter to basal levels after 60 min. | Actin morphology | 28 |
| Colchicine (microtubule destabilizer) | 10 µg/ml | Swiss 3T3 cells, adherent or suspension | Maximal activation of 2-4 fold activation after 30 min | Rhotekin-RBD pulldown | 29 |
| Cytochalasin D (actin filament destabilizer) | 0.5 µg/ml | Swiss 3T3 cells, adherent or suspension | Maximal activation of 2-3 fold after 60 min | Rhotekin-RBD pulldown | 29 |
| Fibronectin (extracellular matrix protein) | Culture plate is coated with fibronectin S | Swiss 3T3 cells | Biphasic regulation after plating cells on fibronectin-coated plates. Initial period of low RhoA activity (10-20 min) followed by a 2-7 fold activation peaking at 60-90 min and then dropping to basal levels after 6 h. | Rhotekin-RBD pulldown | 29 |
Table 3H cont.: Modulators of GTP-Rho levels in cell
| RhoA Modulator | Treatment | Cell type used | Response | Assay Type | Ref. |
|---|---|---|---|---|---|
| Lysophospatidic acid (LPA) (serum lipid & G-protein coupled receptor agonist) | 1 µg/ml | Swiss 3T3 cells, adherent & suspension | Maximal activation of 2-6 fold after 1 min then dropping to basal after 30 min | Rhotekin-RBD pulldown | 29 |
| Lysophospatidic acid (LPA) | 1 µM | N1E-115 neuronal cells | Maximal activation of 3-5 fold after 3 min | Rho-kinase pull down assay | 30 |
| Nocodazole (microtubule destabilizer) | 10 µM | MG63 human osteosarcoma cell & HeLa cells | Maximal activation of 2-3 fold activation after 30 min | Actin morphology& Rhotekin-RBD pulldown | 31 |
| Serum | 5 - 10% | Swiss 3T3 cells, adherent or suspension | Maximal activation of 2-6 fold (10%) & 2-3 fold (5%) after 1-5 min | Rhotekin-RBD pulldown | 29 |
| Sphingosine-1-phosphate (serum lipid & G-protein coupled receptor agonist) | 1 µg/ml | Swiss 3T3 cells, adherent or suspension | Maximal activation of 2-3 fold after 2 min for 3T3 cells and 20 min for HUVEC cells | Rhotekin-RBD pulldown | 29,32 |
| Thrombin (protease, G-protein coupled receptor agonist) | 10 nM | HUVEC human endothelial primary cells | Maximal activation of 14 fold after 2 min, dropped to basal after 30 min | Rhotekin-RBD pulldown | 32 |
| Vinblastine (microtubule destabilizer) | 50 µM | MG63 human osteosarcoma cells | Maximal activation after 30 min | Actin morphology | 31 |
Preparation of lysates from 2D tissue culture cell lines
Activated GTPases are a labile entity and the bound GTP is susceptible to hydrolysis by GAPs during and after cell lysis, resulting in GTPase inactivation. Rapid processing at 4°C is essential for accurate and reproducible results. Detailed instructions for processing lysates from 2D tissue culture cells are given in the G-LISA assay Protocol manuals. The following guidelines are applicable to all G-LISA assays.
1) Wash cells in PBS: Normalization of total cell lysate concentration between samples is required for G-LISA assays. Washing cells in PBS allows removal of protein species from tissue culture media which could interfere with accurate lysate concentration determination.
2) Complete removal of PBS after washing: It is important to maintain the composition of the cell lysis buffer between samples, therefore complete removal of PBS, preferably with an aspirator, should be performed prior to cell lysis.
3) Use cold reagents for processing cells: The PBS used for washing cells and cell lysis buffer should be at 4°C. The cold buffers will help minimize hydrolysis of GTPases during processing. It should be noted, in some cell lines (e.g. 3T3 cells) Cdc42 activation appears to be affected by the temperature of the wash solution. In such cases room temperature PBS may be used. The need to use room temperature PBS should be determined empirically. In all cases, cold lysis buffer supplemented with protease inhibitors should be used.
4) It is recommended to process a “Test plate” prior to the experimental plates. This is used to determine the volume of lysis buffer required to achieve the lysate concentrations recommended in Table 1.
5) Work quickly to process lysates: GTPase proteins are labile entities and will hydrolyze over time. It is recommended to take no more than 15 minutes to process a sample and to process samples sequentially.
6) Snap freeze aliquots of lysate in liquid nitrogen: It is often impractical to process fresh lysates and use them immediately in an activation assay. In particular, if time-points are being taken or multiple samples are being processed, parallel processing is impractical and unnecessary. We highly recommend sequential preparation of samples followed by protein concentration determination, aliquoting and snap freezing aliquots in liquid nitrogen for later analysis. Lysates are stable for several months at -70°C.
7) Keep lysates at 4°C during processing: Lysates should be embedded in ice during processing. This helps to minimize changes in signal over time.
8) Thaw lysates quickly and DO NOT re-freeze: Thawing of cell lysates prior to use in the G-LISA® assay should be in a room temperature water bath, followed by rapid transfer to ice and immediate use in the assay. Transfer to ice while 1/3 of lysate remains frozen as remainder will thaw quickly on ice. Discard any unused lysate.
Preparation of lysates from 3D tissue culture cell lines
Cells grown in Matrigel or collagen gels can be assayed using the G-LISA format. The low amounts of cells in 3D cultures makes the conventional pull-down assay very difficult, variable and expensive.
Keely et al., 2007 (Methods in Enzymology, v426, p27) and Petroll et al., 2008 (J Cell Physiol, v217, p162) give in-depth descriptions using G-LISA technology and 3-D cultures. An additional modification we suggest is to increase the ratio of extract/binding buffer from the recommended 1:1 (v/v) to 1:3
Preparation of lysates from tissue samples
Tissue lysates can be used in G-LISA assays. Recommendations regarding tissue lysates are given below.
1) Ras superfamily GTPases are labile proteins that will hydrolyze bound GTP during sample handling. Tissues should therefore be processed quickly (2 min) and at 4°C. Tissues should be processed immediately using 4°C buffers or cut into small chunks (1 mm diameter), snap frozen in liquid nitrogen, and stored at -70°C for later processing.
2) Tissues can be homogenized using a micro-pestel on ice. Homogenates should be clarified by a 1 minute centrifugation at 4°C. Lysates can be used immediately in an activation assay or snap frozen in “experiment-sized” volumes. We strongly recommend snap-freezing lysates in liquid nitrogen.
3) When possible, tissues should be extracted in the respective G-LISA’s cell lysis buffer as this is the recommended buffer for the respective G-LISA assay (see Table 1).
4) It is recommended that lysis buffer be supplemented with protease inhibitors and phosphatase inhibitors. Recommended inhibitors include: Cytoskeleton protease inhibitor cocktail (Cat# PIC02), sodium fluoride (50 mM final), sodium pyrophosphate (20 mM final), p-Nitrophenyl phosphate (1 mM final), and microcystin LR (1 µM final).
Equalization of Cell Lysate concentrations
Equal protein concentration in all samples is a prerequisite for accurate comparison between samples in GTPase activation assays. Cell extracts should be equalized with ice-cold Lysis Buffer containing protease inhibitor cocktail to give identical protein concentrations. See Table 1 for recommended lysate protein concentrations for each G-LISA. Equalization is not necessary if the variation between samples is less than 10%.
The Precision Red Advanced Protein Assay Reagent, supplied in all G-LISA kits, is a simple one step procedure that results in a red to purple/blue color change characterized by an increase in absorbance at 600 nm.
The assay exhibits low variance in readings between different proteins of the same concentration and high reproducibility of the colorimetric response. The assay can also be performed in approximately 1-2 minutes. These properties are particularly valuable when applied to the labile lysates required for activation assays.
Quick Protein Concentration Method for 1 ml Cuvette (recommended)
Add 20 µl of each lysate or Lysis Buffer into disposable 1 ml cuvettes.
Add 1 ml of Precision RedTM Advanced Protein Assay Reagent to each cuvette.
Incubate for 1 min at room temperature.
Blank spectrophotometer with 1 ml of Precision Red plus 20 µl of Lysis Buffer at 600 nm.
Read absorbance of lysate samples.
Multiply the absorbance by 5 to obtain the protein concentration in mg/ml
Example Calculation
Assume a 20 µl sample of cell lysate added to 1 ml of Precision Red reagent gives an absorbance reading of 0.1.
Where c = protein concentration (mg/ml), A = absorbance reading, l = pathlength (cm), ε = extinction coefficient ([mg/ml]-1 cm-1) and the multiplier of 50 is the dilution factor for the lysate in Precision Red reagent (20 µl lysate in 1 ml Precision Red reagent).
Thus, for a 20 µl sample in 1 ml of Precision Red, the equation becomes C = A x 5
For a 10 µl sample in 1 ml Precision Red, the equation becomes C = A x 10
Quick Protein Concentration Method for 96 Well Plate
- Add 10 µl of each lysate or Lysis Buffer into the well of a 96 well plate.
- Blank spectrophotometer with 290 µl of Precision Red plus 10 µl of Lysis Buffer at 600 nm.
- Add 290 µl of Precision RedTM Advanced Protein Assay Reagent to each well.
- Incubate for 1 min at room temperature.
- Read absorbance of lysate samples.
- Multiply the absorbance by 3.75 to obtain the protein concentration in mg/ml
96 Well Plate Method
The linear range of this assay is 0.05 - 0.4 and is recommended when lysates are below the linear range of the 1 ml cuvette method. The pathlength for 96 well plate readings is 0.8 cm, hence the equation is modified as shown in the example below:
Example Calculation for 96 Well Plate Measurement
Assume a 10 µl sample of cell lysate added to 290 µl of Precision Red reagent gives an absorbance reading of 0.1
Where c = protein concentration (mg/ml), A = absorbance reading, l = pathlength (cm), ε = extinction coefficient ([mg/ml]-1 cm-1) and the multiplier of 30 is the dilution factor for the lysate in Precision Red reagent (10 µl lysate in 290 µl Precision Red reagent).
Thus, for a 10 µl sample in 290 µl Precision Red, the equation becomes C = A x 3.75
For a 5 µl sample in 295 µl Precision Red, the equation becomes C = A x 7.5
NOTE: The protein concentrations generated by the multipliers stated in the method will result in approximate lysate concentrations. Data will be highly reproducible from lysate to lysate and will generate excellent values for relative concentrations of a series of lysates. It should be noted for activation assays, the relative protein concentration between experimental extracts is far more important than the absolute protein quantitation. However, if desired, one can create a standard curve using BSA or IgG protein standards for each experiment. The standard curve should be performed prior to lysate preparations due to the labile nature of the lysates.
Section III: Assay Technical Tips
Use of a multichannel pipettor
When processing more than 16 wells, it is recommended to use a multi-channel or multi-dispensing pipettor with a range of 25 to 200 μl per dispense. Critical steps such as lysate addition, post-binding wash step and the Antigen Presenting Buffer step all have requirements for accurate and timely additions. Attempting to perform >16 assays with a single channel pipettor will also increase the likelihood of allowing wells to dry out before reagent addition can be completed, resulting in variable signals. Therefore, use a multi-channel or a multi-dispensing pipettor wherever possible.
Plate shaker recommendations
It is recommended to use an orbital plate shaker at 400 rpm. As a back-up you can use a 200 rpm orbital shaking culture incubator or a normal orbital rotating platform set to 200 rpm. Signals may be lower with the 200 rpm option. Rocking or tilting plate shakers will not be sufficient for this assay.Below are some examples of acceptable plate shakers:
Model # 4625, Titer Plate Shaker, Lab-Line Instruments, Barnstead Intl.
Model # RF7854, Digital Microplate Shaker, ML Market Lab, researchml.com
Model # RF7855, Incubating Microplate Shaker, ML Market Lab, researchml.com
Vortex of samples after binding buffer addition
Binding buffer is a viscous solution and requires thorough mixing after addition to cell lysates. The best way to do this is a brief 3-5 second vortex on high. This step will greatly reduce variability between duplicate samples and will increase overall assay accuracy.
Spectrophotometer / Luminometer settings
Spectrophotometer: The majority of the work in the design of the colorimetric G-LISA assays has been based on the Molecular Devices SpectraMax 250. The spectrophotometer settings are given in Table 4 as a reference.
Table 4: Spectrophotometer settings for colorimetric G-LISAs
| Parameters | Character | Contents |
|---|---|---|
| Wavelength | 490 nm | Bandwidth 2 nm (can be ± 20 nm for filter based machines) |
| Protocol | End point | Standard end point assay |
| Shaking | Medium, orbital | 5 s |
| Temperature | 24°C | Room temperature is also fine for readings |
Luminometer: Luminometers vary widely in their sensitivity and absolute readings. It is therefore recommended to run a G-LISA assay with blank and positive control to determine that you are in the linear range of the assay. When in the linear range of the assay the positive control should read 4-10 fold higher than the blank wells. Table 5 gives guidelines for luminometer settings;
Table 5: Luminometer settings for luminescense based G-LISAs
| Parameters | Description and Recommendations |
|---|---|
| Gain | Gain controls the sensitivity of the machine. Most luminometers do not allow manual alteration of gain and use an auto-calibration or limited calibration function. It is important to contact the luminometer manufacturer or consult the users manual to determine the best way to alter the machine sensitivity. If gain can be altered one should read at low, medium and high gains to determine the reading within the linear range of the assay (positive control should be 4-7X higher than blank). Gain range varies with instrument, for example gain in the Tecan GmbH SpectroFluor Plus ranges from 0 - 150 (where 150 is the highest). |
| Integration Time | This parameter can be varied on most machines. It is a good idea to set the machine as the lowest integration time (usually 10 – 100 ms). Integration times greater than 200 ms are likely to read out of the linear range of the assay and may require lowering of gain or dilution of primary and/or secondary antibodies (see below). |
| Shaking | Most machines give the shaking option. The recommended setting is 5 s shake, medium orbital speed before read. This option is not essential to the assay. |
| Temperature | Room temperature |
| Plate type | Any setting that specifies 96 well flat, white will be sufficient |
| Filters | Luminescense does not require excitation or emission filters the filter spaces should be left blank. If this is not an option excitation can be set at any value and emission should be set between 400-500nm, with 430-445 as optimal setting. |
Section IV: Data Analysis
Data Analysis: Excel format
1. It is recommended to use the Lysis Buffer wells as reference blanks in all studies with this kit. Based on the operator designating the appropriate wells, most machines have associated protocols that perform this operation automatically. Call Technical Help for the company supplying the plate reader for information on how to perform this function. When the data are “Lysis Buffer subtracted” (Lysis Buffer only samples have been allocated as Blanks in the assay), then you can import them into a simple graph software such as Excel. Alternatively, the Lysis Buffer background can be subtracted manually or in the spreadsheet application.
2. Data should be arranged in columns where the headings are “Sample”, “Mean”, “Standard Deviation”, “rep1”, “rep2”, “rep3” and “rep4” for the number of replicates performed on each sample. E-mail [email protected] or visit cytoskeleton.supremeclients.com to download a free Excel Template.
3. List your samples under the “Sample” column in the same order that they were assayed in the plate.
4. Enter the following formula into the first sector under “Mean”, “=average(Xn:Yn)” where X = the column designator for “rep1”, Y = column designator for “rep4”, and n= row designator of the row that you are working on. Repeat for each sector under the “Mean” header until there are sufficient rows to cover the number of samples in your experiment.
5. Enter the following formula into the first sector under “Standard Deviation”, “=stdev(Xn:Yn)” where X = the column designator for “rep1”, Y = column designator for “rep4”, and n= row designator of the row that you are working on. Repeat for each sector under the “Standard Deviation” header until there are sufficient rows to cover the number of samples in your experiment.
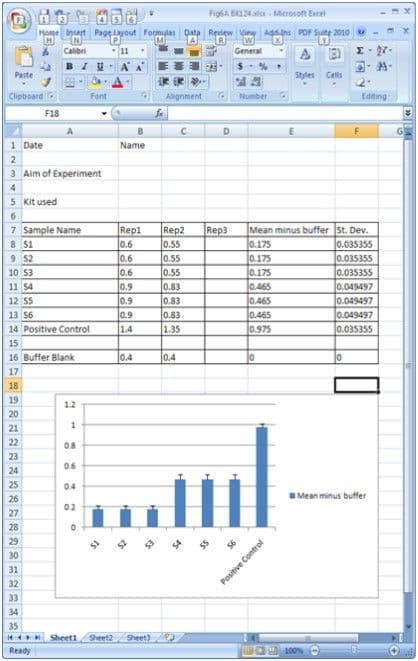
6. Enter your replicate data into rep1, rep2 etc. It doesn’t matter if you only have duplicates because the program will ignore any sectors that do not contain data. The program will calculate the Mean and Standard Deviation of your replicates.
7. When the data has been entered select the Sample, Mean and Standard Deviation data sectors by the click and drag method. Then select the chart making function, in Excel this looks like a clickable square with a mini-bar chart inside. This will guide you through the chart making process with the data you have selected. Choose “column chart” initially, designate the Mean numbers for input values. The Standard Deviation column for the y-axis error bars needs to be designated after the Mean numbers chart is made. This is achieved by double clicking on the graph bars, and selecting the “Y-axis error” tab, then entering the location of the Standard Deviation data by clicking the “Custom” option and selecting the area in the worksheet. E-mail [email protected] or visit cytoskeleton.supremeclients.com to download a free Excel Template.
8. An example of a typical Excel layout and data plot is shown in Figure 1.
Download Experiment Record Sheet
Download Plate Record Template
Technical Assistance: call either 303-322-2254 or e-mail [email protected]
Section V: References
- Niu T.-K. et al. 2005. Dynamics of GBF1, a brefeldin A-sensitive Arf1 exchange factor at the Golgi. Mol. Biol. Cell. 16, 1213-1222.
- Matto M. et al. 2011. Role of ADP ribosylation factor 1 in the regulation of Hepatitus C virus replication. J. Virol. 85, 946-956.
- Tanaka M. et al. 2007. The C-terminus of ephrin-B1 regulates metalloproteinase secretion and invasion of cancer cells. J. Cell Sci. 120, 2179-2189.
- Wessels E. et al. 2006. A viral protein that blocks Arf1-mediated COP-1 assembly by inhibiting the guanine nucleotide exchange factor GBF1. Dev. Cell. 11, 191-201.
- El Azreq M.-A. et al. 2010. Cytohesin-1 regulates the Arf6-phospholipase D signaling axis in human neutrophils: impact on superoxide anion production and secretion. J. Immunol. 184, 637-649.
- Zhang Q. et al. 2007. Small-molecule synergist of the Wnt/b-catenin signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 104, 7444-7448.
- Belov G.A. et al. 2007. Hijacking components of the cellular secretory pathway for replication of poliovirus RNA. J. Virol. 81, 558-567.
- Balasubramanian N. et al. 2007. Arf6 and microtubules in adhesion-dependent trafficking of lipid rafts. Nat. Cell Biol. 9, 1381-1391.
- Goldfinger L. E. et al. 2006. RLIP76(RalBP1) is an R-Ras effector that mediates adhesion-dependent Rac activation and cell migration. J. Cell Biol. 174, 877-888.
- Choi W. et al. 2006. Arf6 plays an early role in platelet activation by collagen and convulxin. Blood. 107, 3145-3152.
- Hafner M. et al. 2006. Inhibition of cytohesins by SecinH3 leads to hepatic insulin resistance. Nature. 444, 941-944.
- Palacios F. & D’Souza-Schorey C. 2003. Modulation of Rac1 and ARF6 activation during epithelial cell scattering. J. Biol. Chem. 278, 17395-17400.
- Sakurai, A. et al. 2010. Semaphorin 3E initiates antiangiogenic signaling through Plexin D1 by regulating Arf6 and R-Ras. Mol. Cell. Biol. 30, 3086-3098.
- Yao et al., 2013. Nonmuscle myosin light-chain kinase mediates microglial migration induced by HIV Tat: Involvement of β1 integrins. FASEB J. doi: 10.1096/fj.12-219600
- Chen et al., 2012. Inhibition of tumor cell growth, proliferation and migration by X-387, a novel active-site inhibitor of mTOR. Biochem. Pharmacol. 83, 1183-1194.
- Romero et al., 2010. Chronic Ethanol Exposure Alters the Levels, Assembly, and Cellular Organization of the Actin Cytoskeleton and Microtubules in Hippocampal Neurons in Primary Culture. Toxicol. Sci. 118, 602-612.
- Kim H.D. et al. 2008. Epidermal growth factor-induced enhancement of glioblastoma cell migration in 3D arises from an intrinsic increase in speed but an extrinsic matrix– and proteolysis-dependent increase in persistence. Mol. Biol. Cell. 19, 4249-4259.
- Tanaka T. et al. 2010. Monocyte chemoattractant proein-1/CC chemokine ligand 2 enhances apoptotic cell removal by macrophages through Rac1 activation. Biochem. Biophys. Res. Commun. 399, 677-682.
- Yang C. et al. 2006. Essential role for Rac in heregulin beta1 mitogenic signaling: a mechanism that involves epidermal growth factor receptor and is independent of ErbB4. Mol. Cell. Biol. 26, 831-842.
- Grill B. & Schrader J.W. 2002. Activation of Rac-1, Rac-2, and Cdc42 by hemopoietic growth factors or cross-linking of the B-lymphocyte receptor for antigen. Blood. 100, 3183-3192.
- Takaya A. et al. 2004. RalA action at nacent lamellipodia of epidermal growth factor stimulated Cos7 cells and migrating madin-darby kidney cells. Mol. Biol. Cell 15, 2549-2557.
- Omerovic J. et al. 2008. Ras isoform abundance and signaling in human cancer cell lines. Oncogene. 27, 2754-2762.
- Satoh T. et al. 1993. Platelet-derived growth factor receptor mediates activation of ras through different signaling pathways in different cell types. Mol. Cell Biol. 13, 3706-3713.
- Perez de Castro I. et al. 2004. Ras activation in Jurkat T cells following low-grade stimulation of the T-cell receptor is specific to N-Ras and occurs only on the Golgi apparatus. Mol. Cell Biol. 24, 3485-3496.
- Scott G.A. et al. 2007. Lysophosphatidylcholine mediates melanocyte dendricity through PKCz activation. J. Invest. Dermatol. 127, 668-675.
- Aguilar H.N. et al. 2011. Phos-tag-based analysis of myosin regulatory light chain phosphorylation in human uterine myocytes. PLoS ONE. 6, e20903.
- Ridley A.J. & Hall A. 1992. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 70, 389-399.
- Schoenwaelder S.M. & Burridge K. 1999. Evidence for a calpeptin-sensitive protein-tyrosine phosphatase upstream of the small GTPase Rho. J. Biol. Chem. 274, 14359-14367.
- Ren X.D. et al. 1999. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 18, 578-585.
- Kranenburg, O. et al. 1999. Activation of RhoA by lysophosphatidic acid and Ga12/13 subunits in neuronal cells: induction of neurite retraction. Mol. Biol. Cell. 10, 1851-1857.
- Zhang, Q. et al. 1997. Lysophosphatidic acid and microtubule-destabilizing agents stimulate fibronectin matrix assembly through Rho-dependent actin stress fiber formation and cell contraction. Mol. Biol. Cell. 8, 1415-1425.
- Vouret-Craviari, V. et al. 2002. Distinct signals via Rho GTPases and Src drive shape changes by thrombin and spingosine-1-phosphate in endothelial cells. J. Cell Sci. 115, 2475-2484.