Introduction
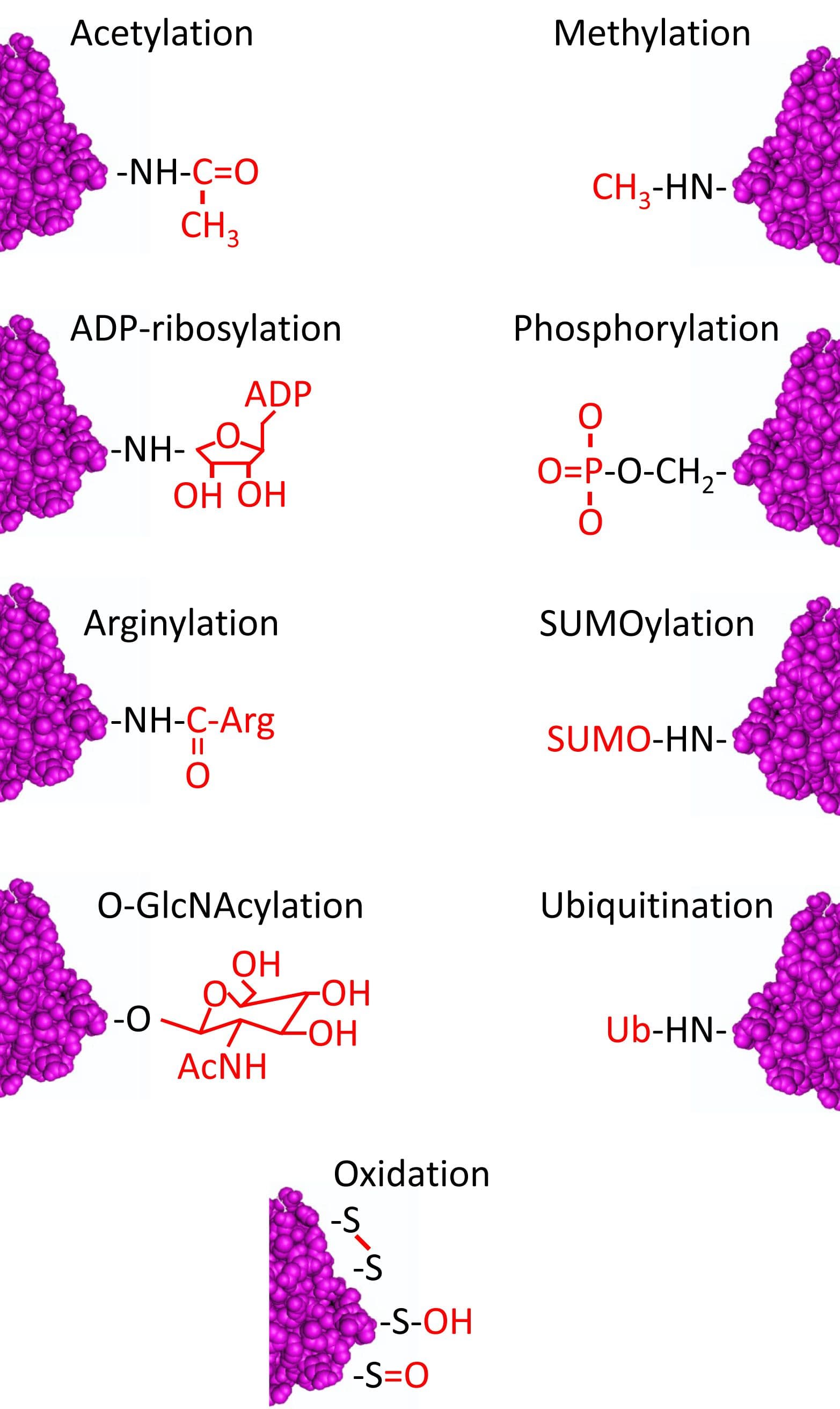
Post-translational modifications (PTMs) are highly dynamic and often reversible processes where a protein’s functional properties are altered by addition of a chemical group or another protein to its amino acid residues (Fig. 1). As a major cytoskeletal protein with roles in cell development, growth, motility, and intracellular trafficking, actin is a major substrate for PTMs. Actin can undergo at least 17 PTMs that include ADP-ribosylation, arginylation, dimethylation, phosphorylation, oxidation, nitration, O-GlcNAcylation, ubiquitination, SUMOylation, acetylation, carbonylation, isaspartylation, transglutamination, malonylation, glutathionylation, nitrosylation, and crosslinking/isopeptide bonding (for review, see ref. 1). Here, we will summarize what is currently known about many of these actin PTMs.
Oxidation PTMs
Reduction-oxidation (redox) reactions constitute a large variety of actin PTMs that include oxidation, nitrosylation, nitration, carbonylation, and glutathionylation. These PTMs occur on many different actin residues including cysteines, methionines, tyrosine, histidine, and tryptophan (Figs. 1 and 9). Actin’s five cysteine residues (Cys374, Cys272, Cys285; Cys217, Cys257) are highly vulnerable to redox modifications. The most vulnerable is Cys374 which is modified by oxidation, glutathionylation, carbonylation, and nitrosylation. PTMs of Cys374 are associated with monomer aggregation via disulfide bonds, decreased polymerization rates, increased critical concentrations, and weakening of actin filaments1. Modifications of other cysteine residues are also linked to reduced polymerization activity and altered binding of actin with actin binding proteins. Similar to cysteines, methionines are also highly vulnerable to redox PTMs and their modification reduces actin’s functionality42,43. Methionines at a variety of positions, including Met44, MET47, Met355, Met176, Met190, Met227, and Met269, are all sites of oxidation. Oxidation of Met176, Met190, and Met269 completely inhibit actin polymerization and induce rapid depolymerization of existing F-actin43.



Oxidation of actin occurs as a result of various signaling pathways throughout the cells of most, if not all, organisms ranging from bacteria to humans. One of the major pathways that mediate actin oxidation is reactive oxygen species (ROS) such as hydrogen peroxide (H2O2) which is produced in response to extracellular signaling as well as during responses to infection or injury44,45. H2O2 inhibits actin polymerization, depolymerizes F-actin, and alters binding between actin and its binding partners44-46.
Actin can also be oxidized directly by enzymes. Mical (Cat. # MIC01), a monooxygenase, oxidizes actin in vivo and in vitro. Mical is activated upon direct binding to F-actin, oxidizing Met44 and Met47 (Cat. # MXA95) to decrease polymerization and depolymerize individual and bundled actin filaments in vivo and in vitro47,48. Active Mical also rearranges F-actin networks, making them more disorganized, creating a chaotic meshwork of short filaments47,48. It is hypothesized that Mical is able to access these “inaccessible” sites with an active site that “fits” between actin monomers48-51.
Other enzymes involved in the oxidation of actin involve the nitric oxide signaling pathway. Two redox PTMs utilizing nitric oxide signaling are nitration and S-nitrosylation. These related PTMs target tyrosine and cysteine residues, respectively, of actin52-58. Nitration (attachment of a nitrite group) on tyrosine amino acid residues leads to the functional impairment of actin and exerts consequences under both normal and pathophysiological conditions58. For example, actin in the liver and kidney cells of sickle cell disease (SCD) patients and animal models of the disease undergoes nitration53. In vivo, three tyrosine residues were nitrated, Tyr91, Tyr198, and Tyr240 as determined by mass spectrometry. In vitro, similar analyses revealed that Tyr53, Tyr198, Tyr240, and Tyr362 are nitrated, though data for Tyr362 were inconsistent53. Functionally, actin nitration reduced the critical concentration, compared to non-nitrated actin, as well as shortening the lag (nucleation) phase and accelerating filament elongation. In human and mouse kidney cells from SCD vs control populations, the altered polymerization dynamics produced F-actin staining that was disorganized and aggregated53. Actin nitration has also been implicated in endothelial barrier dysfunction, an event that can lead to vascular injury and edema54.
S-nitrosylation is a reversible PTM that is also intimately related to nitric oxide signaling and involves the covalent attachment of a nitric oxide group to a cysteine thiol56. Actin’s cysteine residues undergo S-nitrosylation under hyperoxic conditions, increasing the rate of actin polymerization. However, the result is an increased number of short actin filaments55. Hyperoxia-induced actin S-nitrosylation impairs b2 integrin clustering, an important step in responding to infection or injury55. Lu et al. reported that S-nitrosylation of actin also functions in neurotransmission, specifically in controlling neurotransmitter release56 and pain transmission in the spinal cord57. Interestingly, Lu et al.56 found that the S-nitrosylation reduced F-actin content without changing total actin levels. The authors attribute the inhibited release of neurotransmitter to the reduced F-actin content56.
Carbonylation is a form of actin oxidation that results in increased carbonyl content (e.g., aldehyde or ketone groups) of actin. This irreversible PTM leads to cross-linking and aggregation of actin as well as disruption and disassembly of F-actin networks and inhibition of polymerization59-62. Clinically, carbonylation of actin is elevated in the brains of Alzheimer’s disease patients, ischemic hearts, and cell models of inflammatory bowel disease (IBD)61. With IBD, carbonylation of actin compromises the intestinal barrier integrity which disrupts the F-actin network59. Dalle-Donne et al.61 argue that carbonylation is more than just a marker of oxidation; instead indicating severe physiological impairments related to F-actin disruption and disassembly.
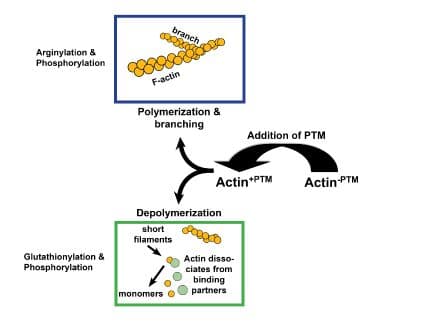
Glutathionylation is another redox PTM that targets two of actin’s cysteine amino acid residues (Cys217, Cys374). Glutathionylation is a reversible PTM whereby glutathione is attached to an actin’s cysteine residue via a disulfide bond, creating glutathione disulfide. Actin glutathionylation serves to protect actin, and thereby cells, from oxidative stress63-65 (Fig. 3). For example, actin glutathionylation is believed to participate in stabilization of axons and dendrites as well as neuron survival during periods of oxidative stress64. Furthermore, actin glutathionylation influences how cells’ actin networks respond to growth factors, mediating actin polymerization and subsequent trafficking and re-arrangement of F-actin66. During oxidative stress, glutathionylation increases which decreases actin polymerization, resulting in reduced F-actin levels1,66 (Fig. 1). Besides inhibiting F-actin formation, increased glutathionylation has also been linked to abnormal rearrangement of actin filaments65,67. Upon reversal of glutathionylation, actin polymerization increases66.
It has also been suggested that actin can act as an antioxidant/scavenger to protect the cell and other proteins from oxidative stressors and oxidative metabolism61,68,69.
Arginylation
Arginylation is mediated by arginyltransferase (Ate1) and involves the addition of arginine on the N-terminus of beta-actin by a peptide bond2,3 (Figs. 1 & 2). Arginylation can impact actin’s function in several ways (Fig. 3). For example, arginylation increases actin polymerization3,4 and strengthens the actin filament network3, the main structural support for maintaining dendritic spine morphology and size5. Blockade/loss of arginylation is associated with defects in cell migration and myofibril contraction4,6-8 as well as collapse of leading edge lamella and reduced F-actin levels3. The leading edge collapse is specifically due to decreased N-terminally arginylated beta actin4. Besides the N-terminus, arginylation also occurs internally on the actin molecule on at least two residues1. Internal arginylation is predicted to affect polymerization and interactions between actin and actin binding proteins1. Ex vivo studies using fibroblasts cultured from Ate1 knockout mice also support a role for arginylation in actin function. Cells have slower rates of polymerization (faster nucleation/slower elongation), decreased F-actin staining, shorter actin filaments, and an increased number of intracellular actin aggregates1,3 (Fig. 1). In vivo, Ate1 knockout mice have defects in cardiovascular development9 and neural crest morphogenesis7.
Acetylation
Actin is irreversibly acetylated on its N-terminus10,11 via the transfer of an acetyl group from acetyl coenzyme A to the termini of alpha-amino groups11 (Figs. 1 & 4). N-terminal acetylation is true for most types of actins12,13. In acetylation, aminopeptidases first remove the N-terminal Met (and sometimes the second amino acid) and then N-terminal acetyltransferases (NATs) modify one or more actin residues. While nonacetylated actin polymerizes and depolymerizes normally12, N-terminal acetylation is required for normal actin structure and complete functionality14,15 (Fig. 3). Specifically, N-terminal acetylation is involved with strengthening actomyosin interactions12-15 and may also influence actin ubiquitination and subsequent processing by the ubiquitin-dependent degradative molecules (for review, see refs 1 & 11).
The N-terminus of actin is not the only site for actin acetylation. At lysine residues away from the N-terminus, histone acetyltransferases (HATs; e.g., PCAF, HDAC6) interact with actin to influence actin binding and function, including transcriptional regulation16.
ADP-Ribosylation
ADP-ribosylation involves the transfer of ADP-ribose from nicotinamide adenine dinucleotide (NAD) to various G-actin residues1,17 (Figs. 1 and 5). ADP-ribosylation is carried out by multiple bacterial toxins and enzymes as well as mammalian enzymes. ADP-ribosylation of G-actin on Arg177 is among the most studied residues. This PTM can be mediated by many different ADP-ribosylating bacterial toxins (e.g., Clostridium botulinum C2)17. ADP-ribosylation at this residue induces cell rounding as well as inhibition of actin polymerization via steric hindrance17. Besides being unable to polymerize, ADP-ribosylated G-actin prevents further polymerization of actin filaments by capping the barbed end of the growing actin filament. ADP-ribosylation at Arg177 also impairs the binding of ATP to actin, actin’s ATPase activity, and nucleation of actin by gelsolin1,17. However, ADP-ribosylation is not strictly a negative regulator of actin dynamics and function17. The bacterial ADP-ribosyltransferase TccC3 produced by Photorhabdus luminescens targets the Thr148 actin residue, leading to increased actin polymerization, possibly due to reduced thymosin-b4 binding17.
An ADP-ribosylactin hydrolase that acts upon ADP-ribosylated actin has been described in mammalian brain, indicating that this PTM is reversible in mammals18. Upon reversal of ADP-ribosylation, F-actin was formed from the actin monomers18.
Cross-linking
Actin monomers can be modified by cross-linking enzymes or chemical reagents on either an intra- or intermolecular level (between monomers of the same filament vs between monomers of adjacent filaments, respectively). Among multiple amino acid residues involved in cross-linking1, some well-studied residues include Gln41 to Lys11319, Gln41 to Cys37420, and Cys374 to Lys19121 (Figs. 1 and 6). These cross-linkings are reported to decrease the speed and force generation of actomyosin without altering binding between actin and myosin or myosin’s ATPase activity22. However, a few studies have reported that actin cross-linking disrupts actomyosin binding and activation of myosin’s ATPase activity23,24. The proteins implicated in cross-linking are bacterial toxins MARTX and VgrG-117 as well as bacterial and eukaryotic transglutaminases1 (TGases). The bacterial toxins induce cross-linking of actin monomers to form polymerization-deficient dimers, trimers, and oligomers17, leading to depolymerization, fragmentation, and proteolytic degradation of F-actin. TGases are enzymes that catalyze an interaction between a glutamine acceptor residue and an amine donor to cross-link actin monomers by themselves, between filaments, or within one filament. TGases can also create actin cross-linkages using polyamine bridges. Although TGase-mediated cross-linking inhibits myosin motor activity (see above), others have reported that TGase-mediated cross-linking has positive effects. For instance, F-actin is stabilized by cross-linking25,26, as is beta-actin in the neuronal actin cytoskeleton27.
Glucose-mediated modifications
Modifications by attachment of polysaccharide chains or glycans to proteins (glycosylation) can occur via multiple mechanisms, including N-linked and O-linked glycosylation, glycosylation with O-linked beta-N-acetylglucosamine (O-GlcNAcylation), and glycation. Typical glucose modifications of actin are by O-GlcNAcylation and glycation as N- and O-linked glycosylation are PTMs characteristic of lumenal proteins and proteins involved in the secretion and trafficking of proteins along membrane-associated pathways1. That is not to say that actin cannot be glycosylated as in utero exposure of rats to alcohol results in glycosylated actin28.
Nuclear and cytoplasmic actin is also modified by O-GlcNAcylation29, a reversible PTM believed to modulate actin’s role in cardiac and skeletal muscle contraction, cardiac health, and diabetes1,30,31 (Figs. 1 and 7). Attachment of O-linked beta-N-acetylglucosamine (O-GlcNAc) onto serine or threonine residues of nuclear and cytoplasmic proteins is controlled by two highly conserved enzymes, O-GlcNAc transferase (OGT) and b-N-acetylglucosaminidase (O-GlcNAcase)32. O-GlcNAc cycles at rates similar to that of O-phosphate in response to various cellular stimuli29,32.
Glycation, or non-enzymatic glycosylation, is another glucose-based modification that results from the non-enzymatic attachment of glucose to lysine residues. Glycation is a PTM linked to diabetes as actin in diabetic patients and animal models of diabetes is susceptible to this PTM33-36. Similar effects of glucose exposure have been reported in cell models of diabetes with decreased activation of myosin ATPase, reduced polymerization, and inhibition of DNAse I36-38.
Methylation
Methylation of actin is an irreversible PTM that occurs on multiple residues, including His7339, which has a part in regulating actin’s interdomain flexibility and stability through stabilization of F-actin (for review, see ref. 1) (Figs. 1 and 8). Another actin residue that undergoes methylation is Lys326 as well as specific actin residues that have been arginylated40. Methylation of arginylated actin residues (a double PTM on the same residue) is likely involved in regulating chromatin structure, gene expression, and nuclear proteins40. Such methylation could also protect arginylated residues from further modification40. Actin methylating enzymes (i.e., protein methylases) have been described39,41, though the exact amino acid residues they target are unknown.
Phosphorylation
Actin has at least 35 amino acid residues that can be modified by phosphorylation and this PTM can exert both negative and positive effects on polymerization (Figs. 1, 3, & 10). For example, actin’s Tyr53 residue in the slime mold Dictyostelium is phosphorylated, which interferes with polymerization, likely through a disruption of actin subunit-subunit contact70,71. Conversely, in the slime mold Physarum, actin fragmin kinase (AFK), a calcium-dependent enzyme, phosphorylates Thr201-203, leading to elongation of the actin filaments72-75. This elongation is believed to be a result of reduced interactions between fragmin and actin. Fragmin is related to the severing protein gelsolin and as such, controls filament length72-75. The effect of Thr phosphorylation is reversed by protein phosphatases PP1 and PP2A76. In both organisms, the changes in actin phosphorylation states are associated with cytoskeletal responses to extracellular events (e.g., locomotion, phagocytosis, signal transduction) and transition into a state of dormancy1,70-73.
In mammals, proteomic analyses have revealed that multiple kinases phosphorylate actin and vary by cell type, disease conditions, and external stimuli. Unfortunately, many of the studies are correlational and do not report a direct relationship between a given kinase and actin phosphorylation1. For example, Ser and Tyr residues on actin are phosphorylated in response to insulin via unknown kinases, leading to reduced DNAse I binding1 (Fig. 1). Likewise, activation of the p21-activated kinase PAK1 leads to actin phosphorylation which is correlated with loss of stress fibers and altered F-actin localization77. Similarly, Src kinase-driven phosphorylation of actin impairs actin polymerization1,78. Several known actin kinases are casein kinase I1,79, cAMP-dependent protein kinase (PKA), and calcium/phosphoinositide-dependent protein kinase (PKC)80,81. Casein kinase I phosphorylates actin similar to AFK (targets Thr and Ser residues and is calcium-dependent). PKA and PKC act in an opposing manner with the former impairing polymerization and the latter stimulating it82,83 (Fig. 1).
Ubiquitination & SUMOylation
Ubiquitination of proteins involves either the addition of one (mono) or more (poly) ubiquitin/ubiquitin-like proteins (Figs. 1 & 11). Actin undergoes both modifications and monoubiquitination is believed to direct subcellular actin localization and confer stability84,85 while polyubiquitination signals actin proteins bound for degradation. Actin is mono- and/or polyubiquitinated by the ubiquitin ligases MuRF1, UbcH5, and Trim3286-89, leading to predictable decreases in actin89. The degradation of ubiquitinated actin is correlated with muscle remodeling and atrophy87-89. The actin binding protein myosin may protect ubiquitinated actin from degradation88. Ubiquitination also serves to degrade actin proteins that have been incorrectly modified by other PTMs such as arginylation90. Besides ubiquitination, actin is also targeted by small ubiquitin-like modifiers (SUMO), which are involved in the nuclear localization of actin91-93 (Figs. 1 and 12). The SUMO proteins are small (approximately 11 kDa) and the modification is covalent and reversible. There are multiple SUMO isoforms and Hofmann et al.93 reported that both SUMO2 and SUMO3 modify nuclear actin through an interaction involving amino acids Lys68 and Lys284.
Summary
Actin is a major cytoskeletal protein whose function is modulated by a variety of PTMs (Figs. 1-12). Despite actin’s relevance in all aspects of cell biology, our current understanding of how at least 17 different PTMs affect actin polymerization, stability, and binding is not complete. As new PTM tools are developed, we can look forward to greatly advancing our understanding of actin PTMs.
References
- J.R. Terman & A. Kashina, 2013. Post-translational modification and regulation of actin. Curr. Opin. Cell Biol. 25, 30-38.
- E. Balzi et al., 1990. Cloning and functional analysis of the arginyl-tRNA-protein transferase gene ATE1 of Saccharomyces cerevisiae. J. Biol. Chem. 265, 7464-7471.
- S. Saha et al., 2010. Arginylation regulates intracellular actin polymer level by modulating actin properties and binding of capping and severing proteins. Mol. Biol. Cell. 21, 1350-1361.
- M. Karakozova et al., 2006. Arginylation of beta-actin regulates actin cytoskeleton and cell motility. Science. 313, 192-196.
- P. Hotulainen et al., 2009. Defining mechanisms of actin polymerization and depolymerization during dendritic spine morphogenesis. J. Cell Biol. 185, 323-339.
- R. Rai et al., 2008. Arginyltransferase regulates alpha cardiac actin function, myofibril formation and contractility during heart development. Development. 135, 3881-3889.
- S. Kurosaka et al., 2010. Arginylation-dependent neural crest cell migration is essential for mouse development. PLoS Genet. 6, e10000878.
- S. Kurosaka et al., 2012. Arginylation regulates myofibrils to maintain heart function and prevent dilated cardiomyopathy. J. Mol. Cell Cardiol. 53, 333-341.
- Y.T. Kwon et al., 2002. An essential role of N-terminal arginylation in cardiovascular development. Science. 297, 96-99.
- J. Vandekerckhove & K. Weber, 1978. Mammalian cytoplasmic actins are the products of at least two genes and differ in primary structure in at least 25 identified positions from skeletal muscle actins. Proc. Natl. Acad. Sci. U.S.A. 75, 1106-1110.
- T. Arnesen, 2011. Towards a functional understanding of protein N-terminal acetylation. PLoS Biol. 9: e1001074.
- A. Abe et al., 2000. Acetylation at the N-terminus of actin strengthens weak interaction between actin and myosin. Biochem. Biophys. Res. Commun. 268, 14-19.
- S. Schmitz et al., 2000. Drosophila ACT88F indirect flight muscle-specific actin is not N-terminally acetylated: a mutation in N-terminal processing affects actin function. J. Mol. Bio. 295, 1201-1210.
- R.K. Cook et al., 1992. Removal of the amino-terminal acidic residues of yeast actin. Studies in vitro and in vivo. J. Biol. Chem. 267, 9430-9436.
- B. Polevoda et al., Nat3p and Mdm20p are required for function of yeast NatB Nalpha-terminal acetyltransferase and of actin and tropomyosin. J. Biol. Chem. 278, 30686-30697.
- A. Obrdlik et al., 2008. The histone acetyltransferase PCAF associates with actin and hnRNP U for RNA polymerase II transcription. Mol. Cell. Biol. 28, 6342-6357.
- K. Aktories et al., 2011. Actin as target for modification by bacterial protein toxins. FEBS J. 278, 4526-4543.
- H. Okamoto et al., 1997. Purification, characterization, and localization of an ADP-ribosylactin hydrolase that uses ADP-ribosylated actin from rat brains as a substrate. J. Biol. Chem. 272, 28116-28125.
- G. Hegyi et al., 1992. Gln-41 is intermolecularly cross-linked to Lys-113 in F-actin by N-(4-azidobenzoyl)-putrescine. Protein Sci. 1, 132-144.
- G. Hegyi et al., 1998. Intrastrand cross-linked actin between Gln-41 and Cys-374. I. Mapping of sites cross-linked in F-actin by N-(4-azido-2-nitophenyl)-putrescine. Biochemistry. 37, 17784-17792.
- M. Elzinga & J.J. Phelan, 1984. F-actin is intermolecularly crosslinked by N,N’-p-phenylene-dimaleimide through lysine-191 and cysteine-374. Proc. Natl. Acad. Sci. U.S.A. 81, 6599-6602.
- E. Kim et al., 1998. Intrastrand cross-linked actin between Gln-41 and Cys-374. III. Inhibition of motion and force generation with myosin. Biochemistry. 37, 17801-17809.
- E. Prochniewicz & T. Yanagida, 1990. Inhibition of sliding movement of F-actin by crosslinking emphasizes the role of actin structure in the mechanism of motility. J. Mol. Biol. 216, 761-772.
- S.D. Duca et al., 2009. Effects of post-translational modifications catalysed by pollen transglutaminase on the functional properties of microtubules and actin filaments. Biochem. J. 418, 651-664.
- E. Kim et al., 1998. Intrastrand cross-linked actin between Gln-41 and Cys-374. II. Properties of cross-linked oligomers. Biochemistry. 37, 17793-17800.
- L. Eli-Berchoer et al., 2000. Effect of intramolecular cross-linking between glutamine-41 and lysine-50 on actin structure and function. J. Muscle Res. Cell Motil. 21, 405-414.
- L. Dolge et al., 2012. Beta-actin is a target for transglutaminase activity at synaptic endings in chicken telencephalic cell cultures. J. Mol. Neurosci. 46, 410-419.
- B. Fofana et al., 2010. Prenatal alcohol exposure alters phosphorylation and glycosylation of proteins in rat offspring liver. Proteomics. 10, 417-434.
- G.W. Hart et al., 2007. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature. 446, 1017-1022.
- 30. S.P. Jones et al., 2008. Cardioprotection by N-acetylglucosamine linkage to cellular proteins. Circulation. 117, 1172-1182.
- G.A. Ramirez-Correa et al., O-linked GlcNAc modification of cardiac myofilament proteins. A novel regulator of myocardial contractile function. Circ. Res. 103, 1354-1358.
- V.V. Lima and R.C. Tostes, 2012. Alternating phosphorylation with O-GlcNAc modification: Another way to control protein function. In: Protein Kinases, Dr. G.D.S. Xavier (Ed.), ISBN: 978-953-51-0640-1. InTech, Rijeka, Croatia.
- W.G. McLean et al., 1992. Posttranslational modifications of nerve cytoskeletal proteins in experimental diabetes. Mol. Neurobiol. 6, 225-237.
- M.R. Brown et al., 1992. Nonenzymatic incorporation of glucose and galactose into brain cytoskeletal proteins in vitro. Neurochem. Int. 21, 177-183.
- C. Pekiner et al., 1993. Glycation of brain actin in experimental diabetes. J. Neurochem. 61, 436-442.
- K.N. Sulochana et al., 2001. Beneficial role of amino acids in mitigating cytoskeletal actin glycation and improving F-actin content: in vitro. Glycoconj. J. 18, 277-282.
- N.V. Kuleva & Z.S. Kovalenko, 1997. Change in the functional properties of actin by its glycation in vitro. Biochem. (Mosc.) 62, 1119-1123.
- H. Resmi et al., 2005. In vitro effects of high glucose concentrations on membrane protein oxidation, G-actin and deformability of human erythrocytes. Cell Biochem. Funct. 23, 163-168.
- M. Raghavan et al., 1992. The use of alternative substrates in the characterization of actin-methylating and carnosine-methylating enzymes. Eur. J. Biochem. 210, 311-318.
- S. Saha et al., 2011. Arginylation and methylation double up to regulate nuclear proteins and nuclear architecture in vivo. Chem. Biol. 18, 1369-1378.
- C. Vijayasarathy & B.S. Rao, 1987. Partial purification and characterization of S-adenosylmethionine:protein-histidine N-methyltransferase from rabbit skeletal muscle. Biochim. Biophys. Acta. 923, 156-165.
- A. Milzani et al., 2000. The oxidation produced by hydrogen peroxide on Ca-ATP-G-actin. Protein Sci. 9, 1774-1782.
- I. Dalle-Donne et al., 2002. Methionine oxidation as a major cause of the functional impairment of oxidized actin. Free Radic. Biol. Med. 32, 927-937.
- A. Milzani et al., 1997. Prolonged oxidative stress on actin. Arch. Biochem. Biophys. 339, 267-274.
- I. Lassing et al., 2007. Molecular and structural basis for redox regulation of beta-actin. J. Mol. Biol. 370, 331-348.
- I. Dalle-Donne et al., 1995. H2O2-treated actin: assembly and polymer interactions with cross-linking proteins. Biophys. J. 69, 2710-2719.
- R.-J. Hung et al., 2010. Mical links semaphorins to F-actin disassembly. Nature. 463, 823-827.
- R.-J. Hung et al., 2011. Direct redox regulation of F-actin assembly and disassembly by mical. Science. 334, 1710-1713.
- J.Q. Guan et al.,2003. Structural reorganization of proteins revealed by radiolysis and mass spectrometry: G-actin solution structure is divalent cation dependent. Biochemistry. 42,11992-12000.
- J.Q. Guan et al., 2005. Structure and dynamics of the actin filament. Biochemistry. 44, 3166-3175.
- K. Takamoto et al., 2007. Biochemical implications of a three-dimensional model of monomeric actin bound to magnesium-chelated ATP. Structure. 15, 39-51.
- S.R. Jaffrey et al., 2001. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat. Cell Biol. 3, 193- 197.
- M. Aslan et al., 2003. Nitric oxide-dependent generation of reactive species in sickle cell disease. Actin tyrosine nitration induces defective cytoskeletal polymerization. J. Biol. Chem. 278, 4194-4204.
- P. Neumann et al., 2006. Peroxynitrite mediates TNF-alpha-induced endothelial barrier dysfunction and nitration of actin. Am. J. Physiol. Lung Cell Mol. Physiol. 290, L674-L684.
- S.R. Thom et al. 2008. Actin S-nitrosylation inhibits neutrophil beta2 integrin function. J. Biol. Chem. 283, 10822-10834.
- J. Lu et al., 2009. Involvement of S-nitrosylation of actin in inhibition of neurotransmitter release by nitric oxide. Mol. Pain. 5, 58.
- J Lu et al., 2011. Rapid S-nitrosylation of actin by NO-generating donors and in inflammatory pain model mice. Mol. Pain. 7, 101.
- M. Aslan, 2012. Functional consequences of actin nitration: in vitro and in disease states. Amino Acids. 42, 65-74.
- A. Banan et al., 2000. Carbonylation and disassembly of the F-actin cytoskeleton in oxidant induced barrier dysfunction and its prevention by epidermal growth factor and transforming growth factor alpha in a human colonic cell line. 46, 830-837.
- I. Dalle-Donne et al., 2001. The actin cytoskeleton response to oxidants: from small heat shock protein phosphorylation to changes in the redox state of actin itself. Free Radic. Biol. Med. 31, 1624-1632.
- I. Dalle-Donne et al., 2003. Protein carbonyl groups as biomarkers of oxidative stress. Clin. Chim. Acta. 329, 23-38.
- J.P. Castro et al., 2012. Carbonylation of the cytoskeletal protein actin leads to aggregate formation. Free Radic. Biol. Med. 53, 916-925.
- I. Dalle-Donne et al., 2005. S-glutathionylation in human platelets by a thiol-disulfide exchange-independent mechanism. Free Radic. Biol. Med. 38, 1501-1510.
- M. Sparaco et al., 2006. Protein glutathionylation in human central nervous system: potential role in redox regulation of neuronal defense against free radicals. J. Neurosci. Res. 83, 256-263.
- E.A. Sabens Liedhegner et al., 2012. Mechanisms of altered redox regulation in neurodegenerative diseases—Focus on S-glutathionylation. Antioxid. Redox Signal. 16, 543-566.
- J. Wang et al., 2003. Stable and controllable RNA interference: Investigating the physiological function of glutathionylated actin. Proc. Natl. Acad. Sci. USA. 100, 5103-5106.
- A. Pastore et al., 2003. Actin glutathionylation increases in fibroblasts of patients with Friedreich’s ataxia. J. Biol. Chem. 278, 2588–42595.
- M.E. Farah et al., 2011. Diverse protective roles of the actin cytoskeleton during oxidative stress. Cytoskeleton. 68, 340-354.
- I. Dalle-Donne et al., 2007. Actin Cys374 as a nucleophilic target of alpha, beta-unsaturated aldehydes. Free Radic Biol. Med. 42, 583-598.
- X. Liu et al., 2006. Phosphorylation of actin Tyr-53 inhibits filament nucleation and elongation and destabilizes filaments. Proc. Natl. Acad. Sci. USA. 103, 13694-13699.
- K. Baek et al., 2008. Modulation of actin structure and function by phosphorylation of Tyr-53 and profilin binding. Proc. Natl. Acad. Sci. USA. 105, 11748-11753.
- B. Constantin et al., 1998. Disruption of the actin cytoskeleton of mammalian cells by the capping complex actin-fragmin is inhibited by actin phosphorylation and regulated by Ca2+ ions. J. Cell Sci. 111, 1695-1706.
- K. Furuhasi & S. Hatano, 1990. Control of actin filament length by phosphorylation of fragmin-actin complex. J. Cell Biol. 111, 1081-1087.
- K. Furuhasi & S. Hatano, 1992. Identification of actin kinase activity in purified fragmin-actin complex. FEBS Lett. 310, 34-36.
- S. Steinbacher et al., 1999. The crystal structure of the Physarum polycephalum actin-fragmin kinase: an atypical protein kinase with a specialized substrate-binding domain. EMBO J. 18, 2923-2929.
- E. Waelkens et al., 1995. Microfilament dynamics: regulation of actin polymerization by actin-fragmin kinase and phosphatases. Adv. Enzyme Regul. 35, 199-227.
- E.A. Papakonstanti & C. Stournaras, 2002. Association of PI-3 kinase with PAK1 leads to actin phosphorylation and cytoskeletal reorganization. Mol. Biol. Cell. 13, 2946-2962.
- C.A. Hirshman et al., 2005. Isoproterenol induces actin depolymerization in human airway smooth muscle cells via activation of an Src kinase and Gs. Am. J. Physiol. Lung Cell Mol. Physiol. 288, L924-L931.
- T. Shibayama et al., 1986. Phosphorylation of muscle and non-muscle actins by casein kinase 1 in vitro. Biochem. Int. 13, 367-373.
- J.M. Carrascosa & O.H. Wieland, 1986. Evidence that (a) serine specific protein kinase(s) different from protein kinase C is responsible for the insulin-stimulated actin phosphorylation by placental membrane. FEBS Lett. 201, 81-86.
- M.P. Walsh et al., 1981. Phosphorylation of smooth muscle actin by the catalytic subunit of the cAMP-dependent protein kinase. Biochem. Biophys. Res. Comm. 102, 149-157
- A.G. Prat et al., 1993. Activation of epithelial Na+ channels by protein kinase A requires actin filaments. Am. J. Physiol. 265, C224-C233.
- Y. Ohta et al., 1987. Protein kinase C and cAMP-dependent protein kinase induce opposite effects on actin polymerizability. FEBS Lett. 222, 305-310.
- S.J. Field et al., 1993. Actin in the merozoite of the malaria parasite, Plasmodium falciparum. Cell Motil. Cytoskeleton. 25, 43-48.
- E. Dantan-Gonzalez et al., 2001. Actin monoubiquitylation is induced in plants in response to pathogens and symbionts. Mol. Plant Microbe. Interact. 14, 1267-1273.
- V. Solomon & A.L. Goldberg, 1996. Importance of the ATP-ubiquitin-proteasome pathway in the degradation of soluble and myofibrillar proteins in rabbit muscle extracts. J. Biol. Chem. 271, 26690-26697.
- C. Polge et al., 2011. Muscle actin is polyubiquitinylated in vitro and in vivo and targeted for breakdown by the E3 ligase MuRF1. FASEB. J. 25, 3790-3802.
- S. Cohen et al., 2009. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J. Cell Biol. 185, 1083-1095.
- E. Kudryashova et al., 2005. Trim32 is a ubiquitin ligase mutated in limb girdle muscular dystrophy type 2H that binds to skeletal muscle myosin and ubiquitinates actin. J. Mol. Biol. 354, 413-424.
- F. Zhang et al., 2010. Differential arginylation of actin isoforms is regulated by coding sequence-dependent degradation. Science. 329, 1534-1537.
- A.C. Vertegaal et al., 2004. A proteomic study of SUMO-2 target proteins. J. Biol. Chem. 279, 33791-33798.
- G. Rosas-Acosta et al., 2005. A universal strategy for proteomic studies of SUMO and other ubiquitin-like modifiers. Mol. Cell. Proteomics. 4, 56-72.
- W.A. Hofmann et al., 2009. SUMOylation of nuclear actin. J. Cell Biol. 186, 193-200.