
VIDEO
+3
Loading...
Cat. #BK121

Kit contents (96 assays: strip-wells allow single or multiple assays per run)
Equipment & materials required
Overview
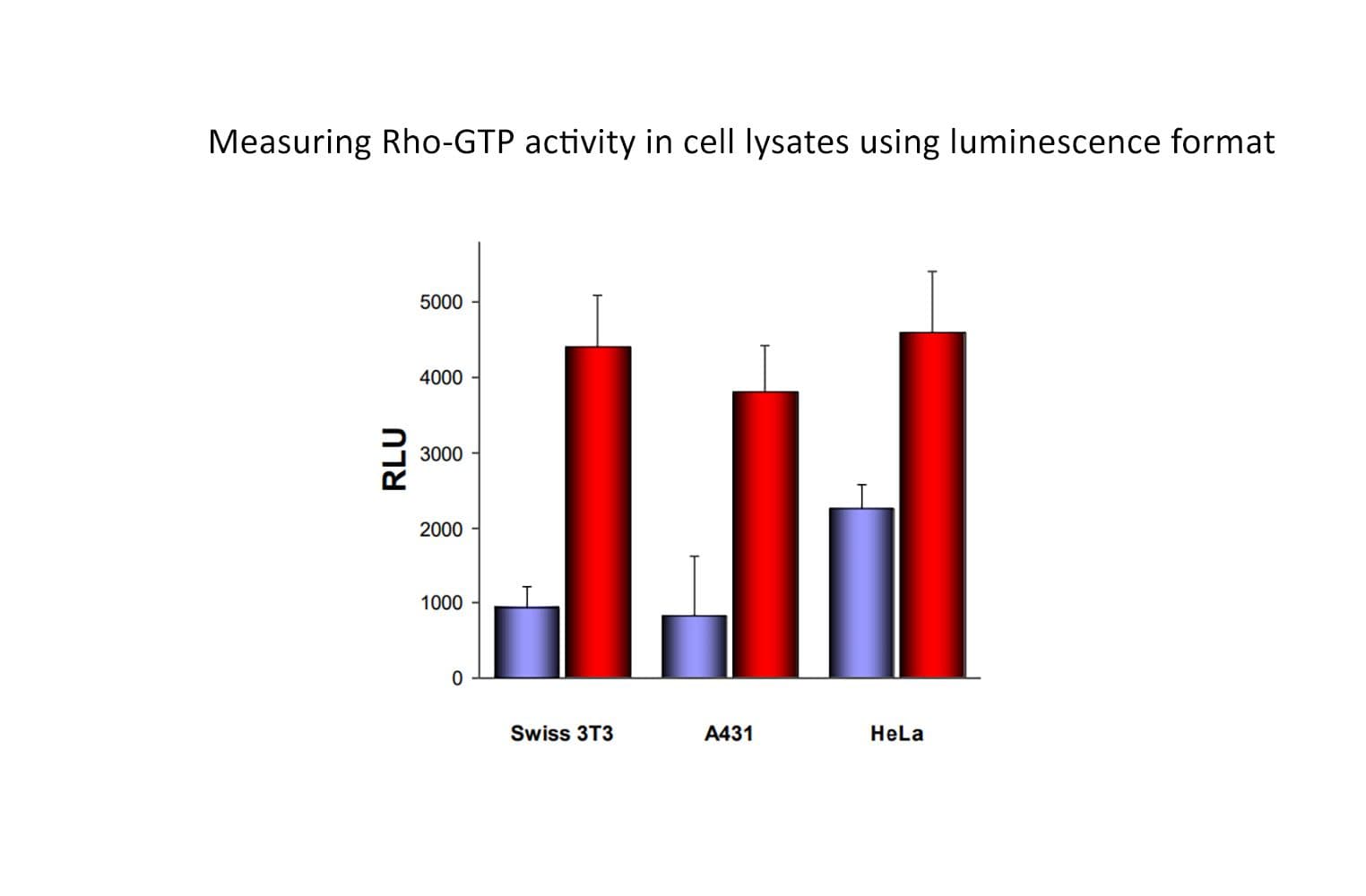
The RhoA G-LISA® assay isolates active RhoA/B/C (GTP-bound form) using the Rho-binding domain (RBD) of an effector protein coupled to a 96-well strip plate. The bound RhoA is then detected and quantified using a luminescence ELISA-based method and a RhoA-specific antibody.
Key characteristics





