Lysosomes are membrane-bound organelles found in nearly all animal cells that were discovered and named by Nobel laureate Dr. Christian de Duve. Lysosomes are spherical vesicles which contain hydrolytic enzymes that can break down virtually all biomolecules (e.g., peptides, nucleic acids, carbohydrates, and lipids). The size of lysosomes varies from 0.1 μm to 1.2 μm. Within the lysosome, an acidic pH of 4.5 - 5.0 is maintained, which is optimal for the activity of the hydrolytic enzymes. The lysosome maintains its pH differential by pumping in protons (H+ ions) from the cytosol across the membrane via proton pumps and chloride ion channels. Vacuolar H+-ATPases are responsible for transport of protons, while the counter transport of chloride ions is performed by ClC-7 Cl−/H+ antiporter. In this way a steady acidic environment is maintained. Lysosomes are known to contain more than 60 different enzymes. The diversity of versatile hydrolytic enzymes is maintained through the importation of enzymes from the Golgi apparatus in small vesicles. The enzymes possess specificity for different substrates; for example, cathepsins are the major class of hydrolytic enzymes, while lysosomal alpha-glucosidase (GAA) is responsible for carbohydrates, and ACP2 is necessary to release phosphate groups of phospholipids. Enzymes of the lysosomes are synthesized in the rough endoplasmic reticulum. Enzymes destined for a lysosome are specifically tagged with the molecule mannose 6-phosphate, so that they are properly sorted into acidified vesicles. The lysosomal membrane protects the cytosol from the degradative enzymes within the lysosome. The cell is additionally protected from any lysosomal acid hydrolases that drain into the cytosol, as these enzymes are pH-sensitive and do not function well or at all in the alkaline environment of the cytosol. This ensures that cytosolic molecules and organelles are not destroyed in case there is leakage of the hydrolytic enzymes from the lysosome.
Besides biomolecular degradation, lysosomes are involved in various cell processes, including secretion, plasma membrane repair, cell signaling, and energy metabolism. The lysosomes also act as the waste disposal system of the cell by digesting unwanted materials in the cytoplasm, both from outside of the cell and from within the cell. Material from the outside of the cell enters through endocytosis, while material from the inside of the cell is digested through autophagy. In addition to being able to break down biomolecules, lysosomes are capable of fusing with other organelles and digesting large structures or cellular debris. For example, through cooperation with phagosomes, they engage in autophagy, clearing out damaged structures. Similarly, they are able to break-down virus particles or bacteria in the phagocytosis of macrophages.
Synthesis of lysosomal enzymes is controlled by nuclear genes. Mutations in the genes for these enzymes are responsible for more than 30 different human genetic diseases, which are collectively known as lysosomal storage diseases (LSDs). These diseases result from an accumulation of specific substrates, due to the inability to break them down. There is no direct medical treatment to cure LSDs. The most common LSD is Gaucher's disease, which is due to deficiency of the enzyme glucocerebrosidase. Consequently, the enzyme substrate, the fatty acid glucosylceramide accumulates, particularly in white blood cells, which in turn affects spleen, liver, kidneys, lungs, brain and bone marrow. The disease is characterized by bruises, fatigue, anaemia, low blood platelets, osteoporosis, and enlargement of the liver and spleen. These genetic defects are related to several neurodegenerative disorders, cancer, cardiovascular diseases, and ageing-related diseases.
Lysosome Images
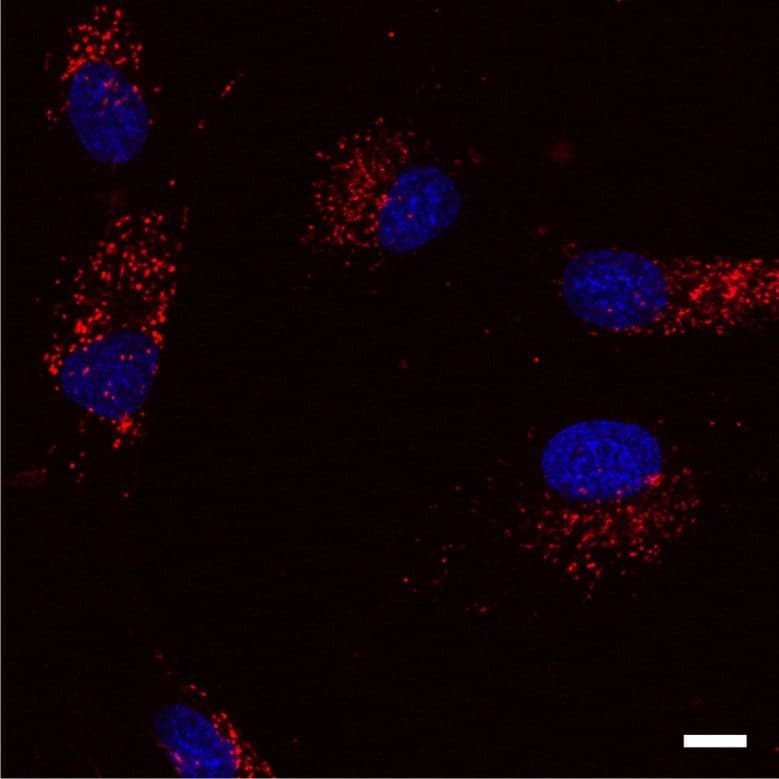
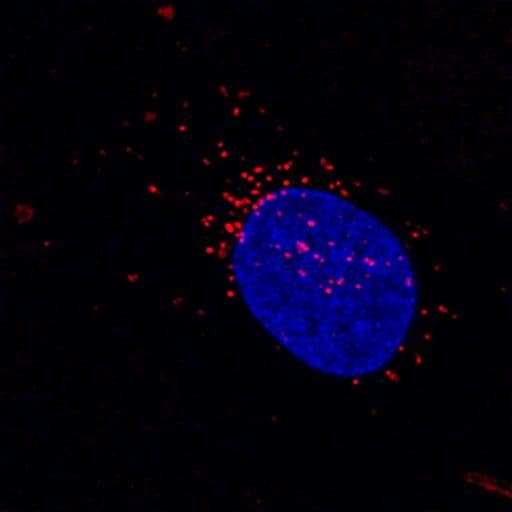
For more information on lysosomes, please see here:
- Mindell J.A. 2012. Lysosomal acidification mechanisms. Annu. Rev. Physiol. 74, 69–86.
- Settembre C. et al. 2013. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 14, 283–296.
- Lüllmznn-Rauch R. 2005. History and morphology of lysosome. In Zaftig P. Lysosomes (Online-Ausg. 1 ed.). Georgetown, Tex.: Landes Bioscience/Eurekah.com. 1, 251-258. ISBN 978-0-387-28957-1.
- Xu H. and Ren D. 2015. Lysosomal physiology. Annu. Rev. Physiol. 77, 57–80.
- Saftig P. and Klumperman J. 2009. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat. Rev. Mol. Cell Biol. 10, 623–635.
- Samie M.A. and Xu H. 2014. Lysosomal exocytosis and lipid storage disorders. J. Lipid Res. 55, 995–1009.
- Platt F.M. et al. 2012. The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J. Cell Biol. 199, 723–734.
- He L.Q. et al. 2013. Autophagy in ageing and ageing-associated diseases. Acta Pharmacol. Sin. 34, 605–611.
- Kuehnel W. 2003. Color Atlas of Cytology, Histology, & Microscopic Anatomy (4th ed.). Thieme. p. 34. ISBN 1-58890-175-0.
- Ishida Y. et al. 2013. A model of lysosomal pH regulation. J. Gen. Physiol. 141, 705–720.
- Parenti G. et al. 2013. New strategies for the treatment of lysosomal storage diseases. Int. J. Mol. Med. 31, 11–20.
- Rosenbloom B.E. and Weinreb N.J. 2013. Gaucher disease: a comprehensive review. Crit. Rev. Oncog. 18, 163–175.
- Sidransky E. 2012. Gaucher disease: insights from a rare Mendelian disorder. Discov. Med. 14, 273–281.
Try these Cytoskeleton Tools for Lysosome Investigation
SiR-Lysosome Kit (50 nmol SiR-Lysosome and 1 µmol verapamil) CY-SC012
SiR700-Lysosome Kit 35 nmol SiR700-Lysosome and 1 µmol verapamil CY-SC016