Key takeaways
- Tau oligomers are soluble, misfolded intermediate structures.
- Tau oligomers are elevated in the frontal cortex of Braak-stage 1 brains.
- Tau oligomers are soluble, misfolded intermediate structures.
- Tau oligomers are elevated in the frontal cortex of Braak-stage 1 brains.
The microtubule-binding protein, Tau, performs an array of functions in neurons, but is notoriously known as an intrinsically disordered protein that becomes dysfunctional and forms neurofibrillary tangles (NFTs) in many neurodegenerative diseases, including Alzheimer’s disease (AD) and other tauopathies(reviewed in 1). AD in particular is a devastating neurological disease that accounts for approximately 60% of senile dementia and is estimated to affect roughly 150 million people by 20502. Due to the presence of Tau NFTs in AD, there has been extensive research on how NFTs may be contributing to the disease and whether therapeutic intervention targeting Tau may reverse AD pathology.
Initial anti-Tau therapies focused on targeting PTMs, inhibiting NFT formation, and stabilizing microtubules, but most have been discontinued due to toxicity or lack of efficacy issues(reviewed in 3). Lack of penetrance through the blood-brain barrier, broad targeting with pan PTM inhibitors, or ineffective targeting of Tau species may all be contributing factors as to why these therapies failed; thus, there has been a renewed focus on more effective targeting strategies.
There is a growing body of research that implicates Tau oligomers, which are structurally distinct from fibrils, as key dysregulated Tau species that contribute to neurodegenerative diseases and may be better therapeutic targets(reviewed in 1, 4).
Early Tau research focused on NFTs and whether disrupting the formation of these structures alleviated neurodegeneration, but some recent studies have pivoted towards investigating Tau oligomers, which are soluble, misfolded intermediate structures(reviewed in 4).While these oligomers are recognized as intermediate or precursor structures for fibril formation5, they have also been shown to play distinct functional roles, including in microtubule binding6, memory impairment7, and cellular uptake(reviewed in 8). Tau oligomers were first identified in AD brain tissue via antibody immunoprecipitation5,9. Importantly, an early study by Maeda et al. showed that Tau oligomers were elevated in the frontal cortex of Braak-stage 1 brains, which is a stage where AD and NFTs are not present; thus suggesting that Tau oligomers may be an early event in AD progression9.
Several studies have linked Tau oligomers to AD progression and pathology. For example, it was shown that Tau oligomers purified from the cerebral cortex of AD brains acted as potent inhibitors of long-term potentiation in hippocampal brain slices and also disrupted memory in wild-type mice10. Another study reported that Tau oligomers from human or mouse sources also impacted LTP and memory, and surprisingly, the impairment occurred rapidly within 20 minutes7. A recent study showed that injecting phosphorylated Tau oligomers into the hippocampus of wild-type mice caused progressive, cognitive defects that were associated with neuronal cell death that spread from the hippocampus to the cortex11. This finding supports data from Colom-Cadena et al., who reported that tau oligomers in AD use trans-synaptic spread in the brain, and are abundant even in areas with low levels of tau fibrils12. Importantly, this report and others showed that the synapse is a critical area for the accumulation of Tau oligomers, and these oligomers are present at higher levels in these synapses relative to phosphorylated Tau, Tau fibrils, and misfolded Tau12,13. Collectively, these findings support the presence of Tau oligomers in AD and suggest that it contributes to disease progression.
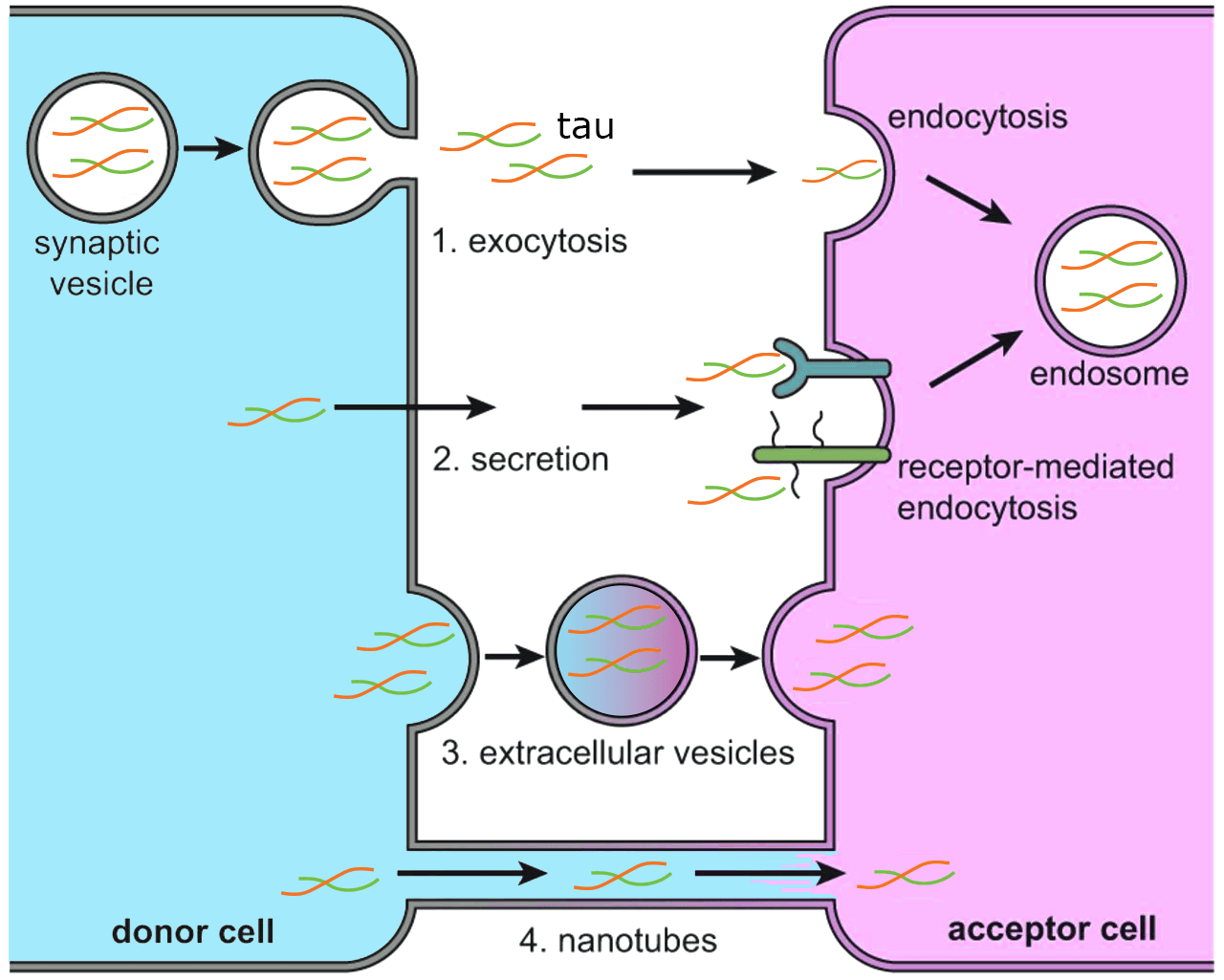
The spread and internalization of Tau by neuronal cells are critical events in the progression of AD(reviewed in 8), and interestingly, Tau oligomers and protofibrils, but not fibrils, isolated from tauopathy mice are internalized by cultured neurons14. Mechanistically, this process involves the generation of transmissible tau seeds, transfer to other cells, and subsequent fiber growth, which has been shown in studies where successive transmission occurred cell to cell, cell to mouse, and from patient samples to mouse brains(reviewed in 1, 4). For example, it was shown that pre-synaptic truncated tau oligomers were released in higher amounts in AD compared to control samples15. Theoretically, Tau oligomers can be transmitted between cells in several ways, including spread through synaptic connections12,16, release of tau into the extracellular space17, exosome release18,19, through endocytic mechanisms20, and via tunneling nanotubes21 (see Figure 1). The current body of information suggests that extracellular Tau appears to be vesicle-free, and can be taken up through binding of extracellular receptors like the low-density lipoprotein receptor-related protein 122,23.
Tau oligomers appear to be more toxic, transmissive, and seeding capable relative to fibrils24,25. How Tau oligomers regulate cellular events to drive neurodegeneration in AD is still under intense investigation, but it was recently shown that Tau oligomers in various neurologic diseases have distinct structural and morphological features, which can differentially affect neuronal function, gene regulation, and disease progression26. Additionally, a recent study investigated Tau oligomer-interacting partners from AD brains versus NDAN (non-demented with Alzheimer’s neuropathology) brains via mass spectrometry and found several unique resilience and vulnerability pathways are altered between the two groups that may shed light on how Tau oligomers contribute to AD27. In one example, the 14-3-3ζ protein was shown to be an important regulator of Tau aggregation and may affect oligomer formation28. When Tau is phosphorylated on S214 and S324, 14-3-3ζ binds Tau and prevents Tau aggregation and condensation at sub-stoichiometric levels. This correlates with two other studies: 1) showed that the heat shock protein 40 (Ydj1) promoted a heterotypic phase separation of Tau that blocked its transition into amyloid fibrils29, and 2) it was recently shown that as Tau forms oligomers, it has a diminished ability to undergo phase separation30. These findings support the premise that there is an important interplay between phase separation and chaperones that regulate Tau aggregation, oligomerization, and fibril formation. The study by Hochmair et al. also reported that upon binding of 14-3-3ζ protein to Tau, the phosphorylated Tau protein dissociates from microtubules28. This may be important as Tau-microtubule interactions are thought to prevent Tau aggregation. This is supported by a recent study that showed tubulin addition into Tau and αSynuclein condensates blocked fibril formation31. These data highlight the importance of identifying key binding partners and regulatory pathways that control Tau aggregation and oligomer formation, which may be especially important in the context of phase separation.
The growing body of work discussed above identifies Tau oligomers as a critical player in AD. The data above make a compelling argument that it may be beneficial to target these oligomers as they are present at early events in AD, they play a critical role in transmission and have detrimental effects in the brain. As therapies that target fibril reduction have not yet shown success, it seems reasonable to also investigate Tau oligomers as viable targets. Several recent studies have begun looking at this(reviewed in 32); for example, calcineurin inhibition has been shown to prevent plasticity deficits induced by brain-derived tau oligomers33, and in another very interesting study, hippocampal neural stem cell-derived exosomes were shown to protect against memory deficits induced by tau oligomers34.
Many products that allow facile entry to this research are provided by Cytoskeleton Inc., including native tau protein, exosome labeling probes (MemGlow), tubulin and MAP fraction, see www.cytoskeleton.com for more information.
1. Van Alstyne, M., J. Pratt, and R. Parker, Diverse influences on tau aggregation and implications for disease progression. Genes Dev, 2025. 39(9-10): p. 555-581.
2. Collaborators, G.B.D.D.F., Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the Global Burden of Disease Study 2019. Lancet Public Health, 2022. 7(2): p. e105-e125.
3. Congdon, E.E., et al., Tau-targeting therapies for Alzheimer disease: current status and future directions. Nat Rev Neurol, 2023. 19(12): p. 715-736.
4. Zheng, H., et al., The Enigma of Tau Protein Aggregation: Mechanistic Insights and Future Challenges. Int J Mol Sci, 2024. 25(9).
5. Maeda, S., et al., Granular tau oligomers as intermediates of tau filaments. Biochemistry, 2007. 46(12): p. 3856-61.
6. Gyparaki, M.T., et al., Tau forms oligomeric complexes on microtubules that are distinct from tau aggregates. Proc Natl Acad Sci USA, 2021. 118(19).
7. Fa, M., et al., Extracellular Tau Oligomers Produce An Immediate Impairment of LTP and Memory. Sci Rep, 2016. 6: p. 19393.
8. Chinnathambi, S., N. Rangappa, and M. Chandrashekar, Internalization of extracellular Tau oligomers in Alzheimer's disease. Adv Clin Chem, 2025. 126: p. 1-29.
9. Maeda, S., et al., Increased levels of granular tau oligomers: an early sign of brain aging and Alzheimer's disease. Neurosci Res, 2006. 54(3): p. 197-201.
10. Lasagna-Reeves, C.A., et al., Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci Rep, 2012. 2: p. 700.
11. Chen, H.R., et al., Intrahippocampal delivery of hyperphosphorylated human tau oligomers induces neurodegeneration in non-transgenic wildtype mice. bioRxiv, 2025.
12. Colom-Cadena, M., et al., Synaptic oligomeric tau in Alzheimer's disease - A potential culprit in the spread of tau pathology through the brain. Neuron, 2023. 111(14): p. 2170-2183 e6.
13. Tai, H.C., et al., Frequent and symmetric deposition of misfolded tau oligomers within presynaptic and postsynaptic terminals in Alzheimer's disease. Acta Neuropathol Commun, 2014. 2: p. 146.
14. Wu, J.W., et al., Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J Biol Chem, 2013. 288(3): p. 1856-70.
15. Sokolow, S., et al., Pre-synaptic C-terminal truncated tau is released from cortical synapses in Alzheimer's disease. J Neurochem, 2015. 133(3): p. 368-79.
16. Pooler, A.M., et al., Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep, 2013. 14(4): p. 389-94.
17. Katsinelos, T., et al., Unconventional Secretion Mediates the Trans-cellular Spreading of Tau. Cell Rep, 2018. 23(7): p. 2039-2055.
18. Saman, S., et al., Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem, 2012. 287(6): p. 3842-9.
19. Tyagi, M., et al., Arc mediates intercellular tau transmission via extracellular vesicles. bioRxiv, 2024.
20. Calafate, S., et al., Loss of Bin1 Promotes the Propagation of Tau Pathology. Cell Rep, 2016. 17(4): p. 931-940.
21. Tardivel, M., et al., Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol Commun, 2016. 4(1): p. 117.
22. Rauch, J.N., et al., LRP1 is a master regulator of tau uptake and spread. Nature, 2020. 580(7803): p. 381-385.
23. Evans, L.D., et al., Tau uptake by human neurons depends on receptor LRP1 and kinase LRRK2. EMBO J, 2025.
24. Lasagna-Reeves, C.A., et al., Preparation and characterization of neurotoxic tau oligomers. Biochemistry, 2010. 49(47): p. 10039-41.
25. Shafiei, S.S., M.J. Guerrero-Munoz, and D.L. Castillo-Carranza, Tau Oligomers: Cytotoxicity, Propagation, and Mitochondrial Damage. Front Aging Neurosci, 2017. 9: p. 83.
26. Lo Cascio, F., et al., Brain-derived tau oligomer polymorphs: distinct aggregations, stability profiles, and biological activities. Commun Biol, 2025. 8(1): p. 53.
27. Jamison, D., et al., Comparative analysis of brain-derived tau oligomer interactomes in Alzheimer's disease, non-demented with Alzheimer's neuropathology, and primary age-related tauopathy: Implications for neurodegeneration and cognitive resilience. J Alzheimers Dis, 2025. 106(4): p. 1486-1508.
28. Hochmair, J., et al., Stoichiometric 14-3-3zeta binding promotes phospho-Tau microtubule dissociation and reduces aggregation and condensation. Commun Biol, 2025. 8(1): p. 1139.
29. Rai, S.K., et al., Chaperone-mediated heterotypic phase separation regulates liquid-to-solid phase transitions of tau into amyloid fibrils. Sci Adv, 2025. 11(23): p. eads1241.
30. Lucas, L., et al., Tau Oligomers Resist Phase Separation. Biomolecules, 2025. 15(3).
31. Lucas, L., et al., Tubulin transforms Tau and alpha-synuclein condensates from pathological to physiological. bioRxiv, 2025.
32. Lin, J., et al., Novel strategies for targeting tau oligomers in neurodegenerative diseases. J Neurol, 2025. 272(6): p. 383.
33. Scaduto, P., et al., Calcineurin inhibition prevents synaptic plasticity deficit induced by brain-derived tau oligomers. Brain Commun, 2024. 6(5): p. fcae277.
34. Krishnan, B., et al., Hippocampal Neural Stem Cell Exosomes Promote Brain Resilience against the Impact of Tau Oligomers. J Neurosci, 2025. 45(16).