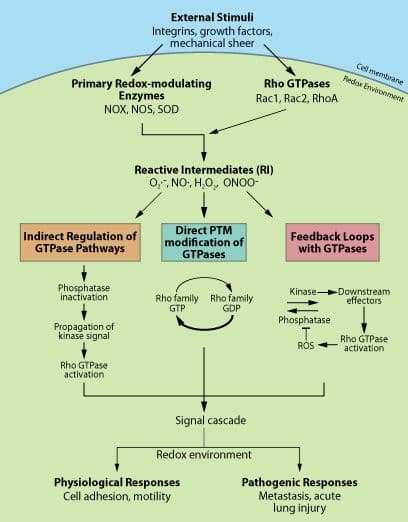
Redox agents, such as reactive oxygen species (ROS) and reactive nitrogen species (RNS), are key regulators in a variety of signal transduction pathways, including integrin signaling, extracellular matrix adhesion, and inflammation1-3. Rho GTPases are also key regulators of many cellular processes, including cell growth, motility, and adhesion4. While redox agents and Rho GTPases operate through a wide array of regulatory mechanisms, crosstalk between ROS/RNS and Rho GTPases is thought to play a pivotal role in many of their physiological functions5,6 and in a growing number of pathological processes such as acute lung injury and cancer7,8. This newsletter broadly summarizes some established and emerging concepts in this rapidy developing field (Fig. 1).

Regulation of ROS by Rho GTPases
Major sources of cellular ROS/RNS production are the primary redox-modulating enzymes such as NADPH oxidases (NOX), nitric oxide synthase (NOS), and superoxide dismutase (SOD)5,9,10 (Fig. 1). Rho GTPases, in particular Rac1 and 2, are a necessary component of activated NOX complexes and the subsequent generation of ROS (superoxide anion, O2.-) from molecular oxygen (O2)11. Upon activation by extracellular signals, the NOX complex assembles at the cell membrane where Rac (GTP-bound and possibly GDI-bound) is required for ROS generation12,13 (Fig. 1). It is not yet clear if Rac acts as an adaptor or mediates electron transfer during O2.- production12. Rac, specifically Rac1, has also been shown to directly interact with NOS enzymes to regulate the generation of nitric oxide (NO)13. The authors found that NOS preferentially binds nucleotide-free GTPase, suggesting the involvement of a guanine nucleotide exchange factor (GEF) and GEF-mediated cellular compartmentalization of Rac-mediated NO generation13. Finally, Rac has been shown to interact directly, in a redox-dependent manner, with SOD, an enzyme that catalyses the conversion of O2.- to hydrogen peroxide (H2O2)14. Under reducing conditions, similar to those in a cell, Rac-GTP binds to SOD, which allows Rac to remain in the active state and propagate the production of O2.- by NOX. Under oxidizing conditions, like those produced locally by O2.- / H2O2 generation, SOD was found to dissociate from Rac which was then converted to its inactive GDP form, possibly by a yet-to-be-identified GTPase activating protein (GAP)14. These reports clearly indicate that Rho GTPases can generate a multitude of reactive oxygen intermediates (RIs) which can, in turn, result in a diverse array of protein modifications. Which species are generated and the cellular responses elicited will depend upon a number of factors, including the given stimuli, the local cell redox potential, and cellular location.
Direct and Indirect Regulation of Rho GTPases by ROS
Recent evidence points towards the interesting possibility that ROS/RNS can directly regulate Rho GTPases through redox-mediated post-translational modifications (PTMs). For example, Heo and Campbell15 demonstrated that the conserved redox sensitive motif Cys18 (Rac numbering) located at the end of the conserved sequence (GXXXXGK(S/T)), found in the p-loop of several Rho GTPases, including RhoA, Rac, and Cdc42, is the site of redox-mediated reversible cysteine oxidation in vitro. This oxidation results in nucleotide displacement and, under reducing conditions, GTP nucleotide exchange and GTPase activation15. This mechanism was later verified in vivo when it was shown that H2O2-mediated activation of RhoA and stress fiber formation is abolished by mutation of the cysteines Cys16 and Cys20 (Rho numbering)16. Importantly, the cysteine mutations do not inhibit the ability of RhoA to be activated by GEFs16. The authors suggested that GTPase activation likely occurs via two parallel mechanisms, classical enzymatic GEFs/GAPs and a redox-mediated exchange (Fig. 1). They postulate that the cells’ redox potential may help determine which mechanism is operational16. Further evidence for a redox-driven exchange mechanism has come from the finding that RhoA is nitrated at Tyr34, within the switch one region of the protein, via enhanced NO production due to acute lung injury7. The authors went on to demonstrate that this PTM produced a GEF-like activation of RhoA. Finally, redox-mediated PTMs have also been proposed to inactivate Rho GTPases17.
In addition to direct regulation, indirect redox-dependent regulation of Rho GTPases involving phosphatases has been reported. For example, Rac-activated ROS production leads to inactivation of RhoA via a mechanism involving redox-mediated inactivation of the low molecular weight protein tyrosine phosphatase (LMW-PTP), subsequent elevation of p190 RhoGAP activity, and deactivation of RhoA18. Interestingly, inactivation of phosphatases by ROS appears to be a common mechanism for ROS regulation of diverse cell pathways19 (Fig. 1).
Feedback Loops between ROS and Rho GTPases
The hypothesis that Rho GTPases participate in redox feedback loops5 has recently been demonstrated for regulation of leukocyte chemotaxis20 (Fig. 1). In a familiar theme, Rac mediates the production of ROS that results in a redox-mediated inactivation of a phosphatase. This in turn allows upregulation of a kinase that mediates GEF activation of Rac20 (Fig. 1). A feedback mechanism has also been proposed for SOD regulation of Rac-mediated generation of ROS14.
Summary
In conclusion, the relationship between Rho GTPases and redox agents is highly complex. Interesting areas for future work might examine the relationship between GAP/GEF-regulated and redox-mediated exchange processes, addressing whether they work in parallel or synergistically, and would they make good druggable targets.
Related Products & Resources
Select Ras & Sos Products
Acetyl Lysine Antibody, Mouse Monoclonal (Cat. # AAC01)
Human SOS1 Protein (Exchange Domain 564-1049) (Cat. # CS-SOS1)
Raf-RBD beads (Binds active Ras protein) (Cat. # RF02)
RhoGEF exchange assay (Cat. # BK100)
G-Switch Activators and Inhibitors
Rho Inhibitor I: ADP ribosylation of Rho Asn-41 (Cat. # CT04)
Rho Activator II:Deamidation of Rho Gln-63 (Cat. # CN03)
Rho/Rac/Cdc42 Activator I (Cat. # CN04)
Rho Pathway Inhibitor I: Rho kinase (ROCK) inhibitor Y-27632 (Cat. # CN06)
Small G-protein Activation Assays and Kits
RhoA Activation Assay Biochem Kit (bead pull down format) (Cat. # BK036)
Rac1 Activation Assay Biochem Kit (bead pull down format) (Cat. # BK035)
Cdc42 Activation Assay Biochem Kit (bead pull down format) (Cat. # BK034)
G-LISA™ RhoA Activation Assay Biochem Kit (colorimetric format) (Cat. # BK124)
G-LISA™ Rac1 Activation Assay Biochem Kit (colorimetric format) (Cat. # BK128)
G-LISA™ Cdc42 Activation Assay Biochem Kit (colorimetric format) (Cat. # BK127)
Activation Assay Video
Cytoskeleton's Small G-protein Activation Assays come in a traditional pull-down bead format or our advanced ELISA based G-LISA format. Learn which Activation Assay format is right for you by watching this video.

References
- Yan B. & Smith J.W. 2000. A redox site involved in integrin activation. J. Biol. Chem. 275, 39964-39972.
- Chiarugi P. et al. 2003. Reactive oxygen species as essential mediators of cell adhesion: the oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. J. Cell Biol. 161, 933-944.
- Leto T.L. & Geiszt M. 2006. Role of Nox family NADPH oxidases in host defense. Antioxid. Redox Signal. 8, 1549-1561.
- Jaffe A.B. & Hall A. 2005. Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 21, 247-269.
- Mitchell L. et al. 2013. Redox regulation of Ras and Rho GTPases: mechanism and function. Antioxid. Redox Signal. 18, 250-258.
- Ferro E. et al. 2014. Ras GTPases are both regulators and effectors of redox agents. Ras Signaling: Methods Mol. Biol. 1120, 55-74.
- Rafikov R. et al. 2014. Lipopolysaccharide-induced lung injury involves the nitration-mediated activation of RhoA. J. Biol. Chem. 289, 4710-4722.
- Myant K.B. et al. 2013. ROS production and NF-kappaB activation triggered by Rac1 facilitate WNT-driven intestinal stem cell proliferation and colorectal cancer initiation. Cell Stem Cell. 12, 761-773.
- Ueyama T. et al. 2006. Involvement of Rac1 in activation of multicomponent Nox-1 and Nox-3 based NADPH oxidases. Mol. Cell Biol. 26, 2160-2174.
- Werner E. 2004. GTPases and reactive oxygen species: switches for killing and signaling. J. Cell Sci. 117, 143-153.
- Hordijk P.L. 2006. Regulation of NADPH oxidases: the role of Rac proteins. Circ. Res. 98, 453-462.
- Bokoch G.M. 1995. Regulation of the phagocyte respiratory burst by small GTP-binding proteins. Trends Cell Biol. 5, 109-113.
- Di-Poi N. et al. 2001. Mechanism of NADPH oxidase activation by Rac/Rho-GDI complex. Biochemistry. 40, 10014-10022.
- Harraz M.M. et al. 2008. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J. Clin. Invest. 118, 659-670.
- Heo J. and Campbell S.L. 2005. Mechanism of redox-mediated guanine nucleotide exchange on redox-active Rho GTPases. J. Biol. Chem. 280, 31003-31010.
- Aghajanian A. et al. 2009. Direct activation of RhoA by reactive oxygen species requires a redox-sensitive motif. PLoS ONE. 4, e8045.
- Zuckerbraun B.S. et al. 2007. Nitric oxide-induced inhibition of smooth muscle cell proliferation involves S-nitrosation and inactivation of RhoA. Am. J. Physiol. Cell Physiol. 292, C824-C831.
- Nimnual A.S. et al. 2003. Redox-dependent downregulation of Rho by Rac. Nat. Cell Biol. 5, 236-241.
- Meng T.C. et al. 2002. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol. Cell. 9, 387-399.
- Kuiper J.W. et al. 2011. Rac regulates PtdInsP3 signaling and the chemotactic compass through a redox-mediated feedback loop. Blood. 118, 6164-6171.