The small G-protein Ras is a GTPase that cycles between inactive (GDP-bound) and active state (GTP-bound) forms. Ras plays an important role in several signal transduction pathways involved in normal cell growth and differentiation1. The three isoforms of Ras, H-Ras, N-Ras, and K-Ras, were identified over 30 years ago for their oncogenic activation in human tumors1,2. Aberrant Ras signaling has been identified in more than 30% of all human cancers with the most common being lung, colon, and pancreatic cancer1,3. In particular, K-Ras has been identified as the most important Ras protein in cancer research, correlated with over 21% of human cancers4. Despite extensive research on these proteins, no effective Ras inhibitor has been identified, earning K-Ras the reputation of an undruggable protein. Several efforts in the past have been unsuccessful, but there may be new hope in developing a therapeutic inhibitor to Ras proteins by targeting the upstream guanine nucleotide exchange factor protein Sos (Son of sevenless). While two human isoforms of Sos exist, Sos1 and Sos2, most research has focused on Sos1. However, not all papers distinguish between Sos1 and Sos2-mediated signaling; in these instances, the term "Sos" is used. Here we discuss several leading concepts in the pursuit of identifying viable drug targets to control atypical Ras signaling.

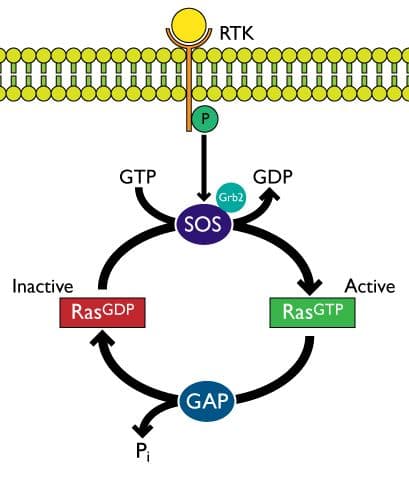
Ras-mediated signaling commonly occurs through the receptor tyrosine kinase (RTK)-Sos-Ras pathway. Dimerization and autophosphorylation of transmembrane RTKs results from binding of growth factor ligands. Sos1 constitutively interacts with growth factor receptor-bound protein 2 (Grb2) and the Sos1/Grb2 complex is recruited to phosphorylated RTKs and binds via the Src homology 2 (SH2) domain of Grb25-9. This RTK/Grb2/Sos1 complex positions Sos to bind to inactive (GDP-bound) Ras located at the plasma membrane (and intracellular membranes)5. Binding of Sos to Ras activates Ras by facilitating the exchange of GDP for GTP. Active GTP-bound Ras is returned to its inactive GDP-bound state through the activity of GTPase activating proteins (GAPs) (Figure 1)1,6.
Although Sos-Ras interactions were described two decades ago7, recent studies have identified new binding pockets, small molecules, and important post-translational modifications that may lead to the development of novel Ras inhibitors that work by disrupting Sos-Ras binding. Four recent papers describing the exciting results of altering Sos-Ras interactions are briefly described here. (1) Peptide HBS3 (designed and synthesized by the hydrogen bond surrogate [HBS] approach) mimics the alpha helix of Sos that is inserted into the switch region of Ras during Sos-Ras binding. In vitro experiments utilizing the alpha-helical mimic interfered with Sos-Ras binding and resulted in significant reduction in Ras activation10; (2) A unique ligand-binding pocket between the switch I/II regions of Ras has been identified and the small molecule DCAI, designed by fragment-based lead discovery (FBLD), specifically binds within this pocket and blocks the interaction between Sos1 and K-Ras, resulting in K-Ras inhibition11; 3) SUMOylation of Grb2 at Lysine 56 located in the linker region increases the formation of Sos1/Grb2 complexes, which leads to ERK/MAPK activation, presumably mediated by Ras. Conversely, inhibition of SUMOylation of Grb2 at Lysine 56 impairs Sos1-Grb2 binding12; (4) K-Ras is post-translationally acetylated at Lysine 104 which negatively regulates K-Ras by interfering with Sos1-mediated nucleotide exchange. Modulating the acetylation state of K-Ras via activating its acetyltransferase or inhibiting its deacetylase may lead to cancer therapeutics13.
An ideal therapeutic K-Ras inhibitor will selectively inhibit mutant K-Ras. Mutant vs wildtype selectivity has proved challenging since mutations are distal from the effector binding sites1. Single amino acid mutations of K-Ras at residues 12, 13, and 61 are common and render K-Ras a constitutively active (GTP-bound) GTPase4. Dr. Kevan Shokat and his team at the Howard Hughes Medical Institute at the University of CA, San Francisco, recently made progress in addressing this selectivity issue by identifying a new allosteric switch II pocket (S-IIP) in a specific type of K-Ras mutant (G12C; Glycine 12 to Cysteine), which is present in 7% of lung cancer patients14. Small molecules were designed to bind to this mutated Cysteine and disrupt switch I and switch II regions. As a result, Sos-mediated nucleotide exchange is compromised and mutant K-Ras inhibition is achieved.
As researchers reveal additional Ras regulatory mechanisms, sensitive and quantitative methods to measure activated levels of these GTPase proteins take on increasing importance. To aid researchers with identifying and characterizing cancer therapeutics, Cytoskeleton, Inc. offers many small G-protein tools such as pull-down and G-LISA activation assays which are widely used to measure the activated levels of small G-proteins. Pull-down methods measure activated GTPase levels utilizing a domain of an effector protein coupled to a bead and the activated level of protein is measured by Western blot. G-LISA activation assays are faster, more sensitive, and more quantitative than traditional pull-down techniques. A video outlining these methods is available at cytoskeleton.supremeclients.com/activationassayvideo. Small G-protein antibodies, activators, and inhibitors are also available to help elucidate Ras activation pathways.
Related Products & Resources
Select Ras & Sos Products
Acetyl Lysine Antibody, Mouse Monoclonal (Cat. # AAC01)
Human SOS1 Protein (Exchange Domain 564-1049) (Cat. # CS-SOS1)
Raf-RBD beads (Binds active Ras protein) (Cat. # RF02)
RhoGEF exchange assay (Cat. # BK100)
G-Switch Activators and Inhibitors
Rho Inhibitor I: ADP ribosylation of Rho Asn-41 (Cat. # CT04)
Rho Activator II:Deamidation of Rho Gln-63 (Cat. # CN03)
Rho/Rac/Cdc42 Activator I (Cat. # CN04)
Rho Pathway Inhibitor I: Rho kinase (ROCK) inhibitor Y-27632 (Cat. # CN06)
Small G-protein Activation Assays and Kits
Ras Activation Assay Biochem Kit (bead pull down format) (Cat. # BK008)
RhoA Activation Assay Biochem Kit (bead pull down format) (Cat. # BK036)
Rac1 Activation Assay Biochem Kit (bead pull down format) (Cat. # BK035)
Cdc42 Activation Assay Biochem Kit (bead pull down format) (Cat. # BK034)
G-LISA™ Ras Activation Assay Biochem Kit (colorimetric format) (Cat. # BK131)
G-LISA™ RhoA Activation Assay Biochem Kit (colorimetric format) (Cat. # BK124)
G-LISA™ Rac1 Activation Assay Biochem Kit (colorimetric format) (Cat. # BK128)
G-LISA™ Cdc42 Activation Assay Biochem Kit (colorimetric format) (Cat. # BK127)
Activation Assay Video
Cytoskeleton's Small G-protein Activation Assays come in a traditional pull-down bead format or our advanced ELISA based G-LISA format. Learn which Activation Assay format is right for you by watching this video.

References
- Wang W. et al. 2012. Ras inhibition via direct Ras binding—is there a path forward. Bioorg. Med. Chem. Lett. 22, 5766-5776.
- Castellano E. and Santos E. 2011. Functional specificity of Ras isoform: so similar but so different. Genes Cancer. 2, 216-231.
- Burns M.C. et al. 2014. Approach for targeting Ras with small molecules that activate SOS-mediated nucleotide exchange. Proc. Natl. Acad. Sci. U.S.A. 111, 3401-3406.
- Baines A. et al. 2011. Inhibition of Ras for cancer treatment: the search continues. Future Med. Chem. 3, 1787-1808.
- McKay M.M. and Morrison D.K. 2007. Integrating signals from RTKs to ERK/MAPK. Oncogene. 26, 3113-3121.
- Innocenti M. et al. 2002. Mechanisms through which sos1 coordinates the activation of Ras and Rac. J. Cell Biol. 156, 125-136.
- Bar-Sagi D. 1994. The Sos (Son of sevenless) protein. Trends Endocrinol. Metab. 5, 165-169.
- Ceresa B.P. and Pessin J.E. 1998. Insulin regulation of the Ras activation/inactivation cycle. Mol. Cell. Biochem. 182, 23-29.
- Lowenstein E.J. et al. 1992. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell. 70, 431-442.
- Patgiri A. et al. 2012. An orthosteric inhibitor of the Ras-Sos interaction. Nat. Chem. Biol. 7, 585-587.
- Maurer T. et al. 2012. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc. Natl. Acad. Sci. U.S.A. 109, 5299-5304.
- Qu Y. et al. 2014. SUMOylation of Grb2 enhances the ERK activity by increasing its binding with Sos1. Mol. Cancer. 13, 95
- Yang M.H. et al. 2012. Regulation of RAS oncogenicity by acetylation. Proc. Natl. Acad. Sci. U.S.A. 109, 10843-10848.
- Ostrem J. et al. 2013. K-Ras (G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 503, 548-551.
*Acknolegments:
We thank Dr. Marina Pasca di Magliano and Meredith A. Collins for providing the header image on page 1. Similar images are described in detail here: J Clin Invest. 2012;122(2):639–653. doi:10.1172/JCI59227.