SUMOylation of a target protein is a recently discovered post-translational modification that has been shown to play a critical role in protein structure and function. Recent proteomic studies have identified hundreds of proteins that are SUMOylated. As SUMO modifications are relatively new compared to other PTMs, tools to investigate SUMO are not well established. Appropriate lysis buffers, high quality affinity reagents, and appropriate inhibitors are all critical reagents for effectively studying SUMO modifications. Here we describe a newly developed Signal-Seeker SUMO 2/3 detection kit (BK162) that rapidly identifies SUMO 2/3 modifications for any target protein. This kit utilizes optimized buffers and inhibitors to preserve SUMO 2/3 modifications unlike conventional, non-denaturing lysis buffers. Additionally, the sensitivity of the kit allows for investigation of endogenous and dynamic changes of SUMO 2/3 modifications. Finally, we show that the (BK162) kit allows for SUMO 2/3 detection from both cell culture and tissue. Collectively, these studies highlight the utility of the (BK162) kit to investigate SUMO 2/3 modifications of several target proteins in various applications.


Introduction
Dynamic post translational modifications (PTMs) like small ubiquitin- modifier (SUMO)ylation provide proteins with vastly expanded functionality. SUMO modifications regulate many biological process, such as, transcriptional regulation and DNA repair1. In vertebrates 4 paralogs of SUMO exist, SUMO-1, SUMO-2, SUMO-3, and SUMO-42. SUMO-2 and SUMO-3 are referred to as SUMO 2/3 because of their high similarity, and the inability to distinguish the two molecules endogenously. Of interest, SUMO-2/3 appears to be more abundant than SUMO-1 and there is a free pool of SUMO-2/3 molecules that are utilized in response to cellular stresses3. Recent proteomic studies have identified hundreds of proteins that are SUMOylated4,5. Still, these data need complementary validation and mechanistic studies to better define the importance of a given protein’s SUMO state both physiologically and pathologically.
As SUMO modifications are relatively new compared to other PTMs like ubiquitination or acetylation, tools to investigate SUMO are not well established. Appropriate lysis buffers, high-quality affinity reagents, and appropriate inhibitors are all critical reagents for effectively studying PTMs; in particular, SUMO modifications. Cytoskeleton’s SUMO 2/3 antibodies have previously been validated for WB, IP and IF applications (See Validation of SUMO-2/3 Antibodies White Paper). Here, we provide tips for investigating target protein SUMOylation while utilizing Cytoskeleton’s SUMO 2/3 detection kit (BK162).
Results and Discussion
Importance of lysis buffers and inhibitors when investigating SUMO 2/3 modifications
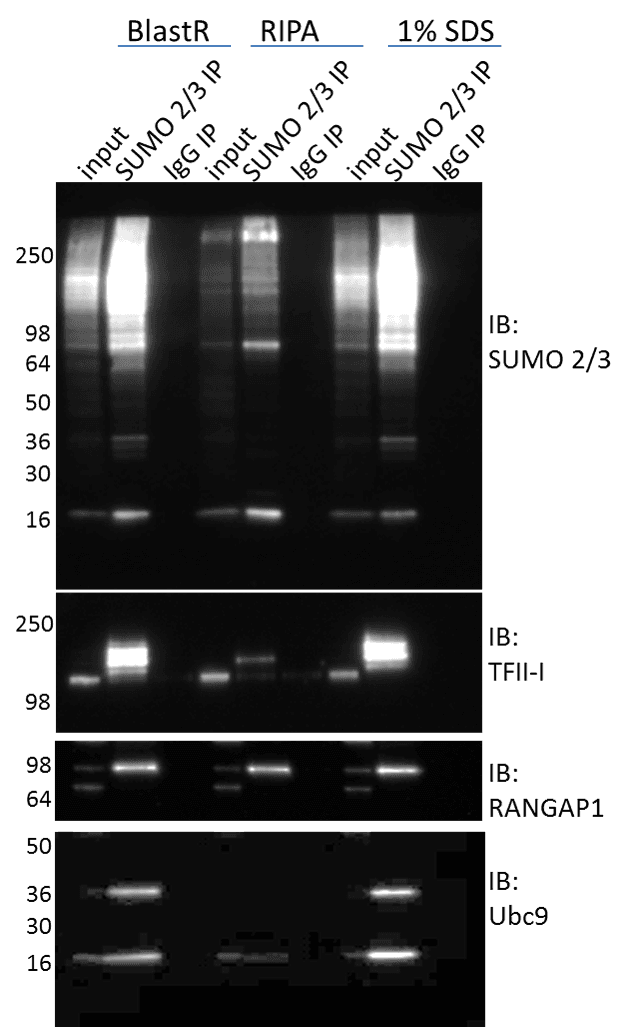
SUMO isopeptidases (deSUMOylases) can maintain a high level of activity in a variety of lysis buffers (e.g. RIPA), which results in loss of the SUMO modification6. This loss of SUMO signal makes detecting SUMO modified proteins challenging. One approach to inactivating deSUMOylases is to utilize denaturing lysis buffers. Figure 1 shows immunoprecipitation (IP) enrichment of total SUMO 2/3 profiles, as well as, target specific SUMO 2/3 modified proteins isolated from denaturing (BlastR and 1% SDS) versus non-denaturing (RIPA) lysis buffers. The total SUMO 2/3 signal is clearly reduced in non-denaturing lysate, and supports the established dogma whereby utilizing denaturing lysis buffers are necessary to preserve the SUMO 2/3 signal. When investigating target specific SUMO 2/3 modifications like TF-II or Ubc9, the signal is nearly undetectable when utilizing RIPA buffers.
Importantly, Cytoskeleton’s recently developed lysis buffer, BlastR, was as effective as 1% SDS denaturing buffer for preserving SUMO 2/3 modifications (Figure 1). The BlastR buffer system has several additional benefits compared to 1% SDS; specifically, easy protein quantitation using standard colorimetric assays, simplified genomic DNA removal using the BlastR filter, and a high compatibility with affinity reagents (see BlastR Rapid Lysate Prep Kit White Paper for more information).
The high activity of deSUMOylases in cell lysates are also inhibited using the deSUMOylase inhibitor, NEM. The (BK162) kit utilizes NEM in combination with BlastR buffer to maximize SUMO 2/3 enrichment by minimizing signal-loss due to deSUMOylase activity. Figure 2 clearly shows that lysing cells without NEM results in significant deSUMOylation of all proteins, as exemplified by loss of total and target-specific SUMO 2/3 signal. These data collectively highlight the importance of choosing the appropriate lysis buffer and inhibitors when studying SUMOylation.
Additionally, data in figure 2 identify a simple approach for validating a SUMO 2/3 modification signal. Simply lysing cells with or without NEM allows the investigator to determine if the SUMO 2/3 signal is real, as the band should disappear when NEM in removed from the lysis buffer. While additional validation methods should also be performed, this experiment may provide additional confidence in a SUMO 2/3 result.

Investigating endogenous changes in SUMO 2/3 modifications requires robust enrichment and detection systems
A second drawback when investigating SUMO 2/3 modifications is that only a small but critical fraction of a target protein’s population is SUMO modified6. Thus, investigating whether a protein is modified is often carried out using overexpression systems5. While this system can provide critical information about whether a protein is SUMO 2/3 modified or not, it can also interfere with interpretation of the target protein’s physiologic SUMO 2/3 state under specific conditions. To effectively understand the mechanistic role of SUMO 2/3 modification for a protein of interest, endogenous investigation of the target protein’s SUMO 2/3 state should be investigated.
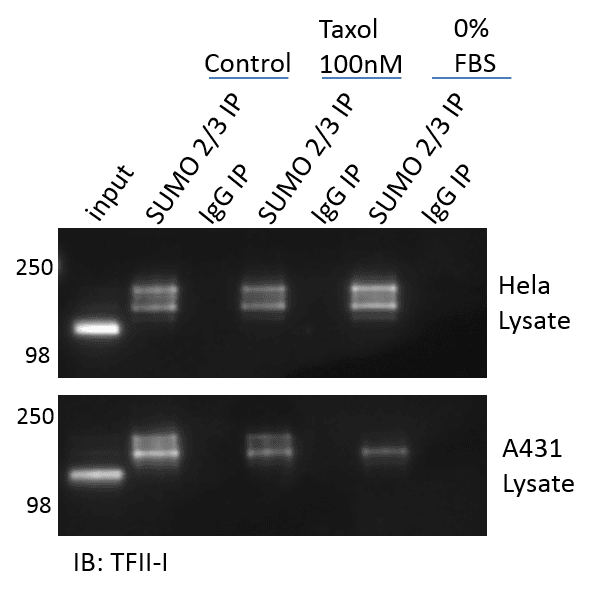
Figure 3 provides results obtained using the (BK162) kit (BlastR lysis, ASM24-beads, NEM, etc..) to effectively enrich and detect endogenous TFII-I changes in response to both taxol treatment as well as serum depletion (0% FBS). The data highlights the ability of the (BK162) kit to detect modest, but endogenous changes in TFII-I SUMO 2/3 modifications in response to different stimuli, which alter the SUMO 2/3 state in both a positive and negative direction. Interestingly, the stimulant Taxol reduces TFII-I’s SUMO 2/3 state in both Hela and A431 cells. These changes appear modest, but were highly reproducible. It is fair to wonder whether these changes would be detectable in an overexpression system, or would the signal be completely masked? Removal of FBS from the cell culture media for 24 hours increased the TFII-I SUMO 2/3 state in Hela cells, but reduces it in A431 cells. These data highlight the variability in PTM results when using different cell line models, and highlights the importance of having tools to detect endogenous SUMO 2/3 modifications in multiple model systems.
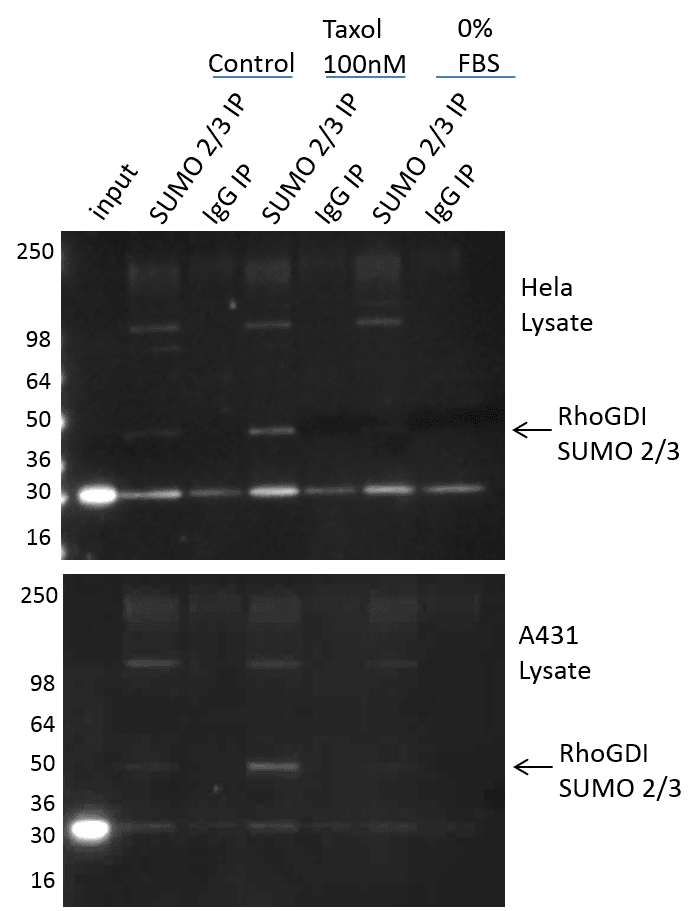
The (BK162) kit works very well with a protein like TFII-1 that is highly modified by SUMO 2/3, but the question remained on how well it could detect SUMO 2/3 endogenous changes on a protein that is not abundantly modified by SUMO 2/3. RhoGDI was chosen because it has been shown to be SUMO 2/3 modified in response to taxol, but only with an overexpression system7. In fact, the author’s stated that they were unable to detect SUMO 2/3 modified RhoGDI endogenously. Figure 4 shows RhoGDI SUMO 2/3 modification in response to taxol and serum restriction. The (BK162) kit effectively isolates SUMO 2/3 modified RhoGDI (prominent band below 50 kDa marker; highlighted arrow). This SUMO 2/3 identification was performed on endogenous RhoGDI in both A431 and Hela cells, and was highly reproducible. Interestingly, serum restriction appears to drive down the already low SUMO 2/3 modified RhoGDI.
These findings highlight the importance of using robust SUMO 2/3 affinity reagents that have been optimized for enriching and detecting endogenous SUMO 2/3 modified target proteins. Additionally, the (BK162) kit also provides IgG control beads (CIG01), which allows the user to determine the specificity of enrichment, and will often times highlight the unmodified protein on interest when it binds non-specifically (see RhoGDI, Figure 4).
Investigation of SUMO 2/3 modified proteins from tissues
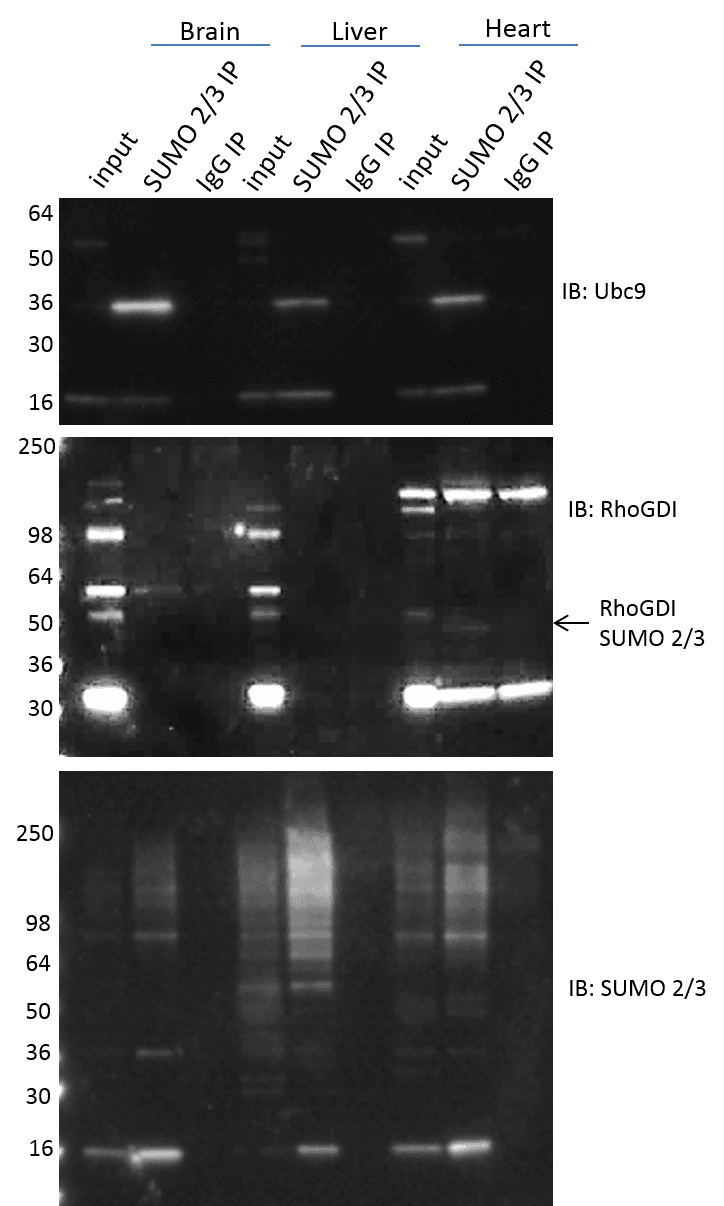
Investigation of SUMO 2/3 modified proteins in tissue is often desirable to translate findings from cell culture to animal models, as well as when investigating mechanistic causes of specific disease models. The (BK162) effectively isolates and enriches SUMO 2/3 modified proteins in both cell culture and tissue lysates. Mouse brain, liver, and heart were isolated, and the tissue was processed with the BlastR lysis system (see methods). SUMO 2/3 modified proteins were enriched from the various tissue lysates using ASM24-beads and the results show effective enrichment of total and specific SUMO 2/3 modified proteins (Figure 5). It is interesting to see the high variability in SUMO 2/3 signals between different tissue types. Note, that the SUMO 2/3 signal intensity for a specific protein (i.e. Ubc 9) does not correlate with the signal intensity observed for the total SUMO 2/3 protein profile. These data highlights the importance of investigating both total as well as target protein specific SUMO 2/3 changes. It was also intriguing to find specific SUMO 2/3 modification of RhoGDI in heart tissue, but not brain or liver in wild type mice. It will be interesting to determine if this is mechanistically important, in regulating RhoGDI in heart versus other tissue types. Additionally, it will be interesting to determine if the RhoGDI SUMO 2/3 state is activated in brain and liver in certain disease models.
Summary
The (BK162) SUMO 2/3 detection kit provides a comprehensive toolset to rapidly discover endogenous SUMO 2/3 modification for target proteins. This system maximizes SUMO 2/3 detection by diminishing the pitfalls that are normally encountered when investigating SUMO 2/3 modifications. This allows users to identify novel SUMO 2/3 modifications, as well as investigate dynamic endogenous SUMO 2/3 changes that may be critical for mechanistic regulation. Importantly, this system allows investigators to study SUMO 2/3 modifications in both cell and tissue using the same system to simplify investigation
Materials and Methods
Cell Culture and Reagents
A431 and Hela cells were grown in DMEM media (ATCC, VA) supplemented with 10% FBS (Atlas Biologicals, CO) and penicillin/streptomycin (ThermoFisher, MA). Trypsin/EDTA was obtained from Gibco (ThermoFisher, MA). Unless otherwise noted, chemicals were obtained from Sigma Chemical Co. (Sigma, MO). Paclitaxel (taxol) (TXD01) was obtained from Cytoskeleton, Inc. (Cytoskeleton, CO). For taxol stimulation experiments A431 and Hela cells were plated at 50% confluency and allowed to recover for 48hrs. The cells were then treated with 100 nM taxol for 6 hours. For serum restriction experiments the cells were plated at 50% confluency and allowed to recover for 24 hours. The media was changes to DMEM without FBS for a 24 hour treatment. Experiments were performed in 15cm dishes (Corning, NY).
Western blotting
Untreated or treated cells were lysed with ice-cold BlastR lysis (Cytoskeleton, CO) containing a cocktail of NEM and protease inhibitors (PIC02) (Cytoskeleton, CO) unless specifically stated. DNA was removed by passing the lysate through the BlastR filter system (Cytoskeleton, CO). After dilution with BlastR dilution buffer, protein concentrations were determined with protein reagent, ADV02 (Cytoskeleton, CO), and measured at 600nm OD. Protein lysate samples were separated using Tris-glycine SDS-polyacrylamide gel electrophoresis (ThermoFisher, MA) and transferred to Immobilon- P membranes (Millipore, MA). Membranes were blocked for 1 hr at room temperature in Tris-buffered saline (10 mM Tris-HCl, pH 8.0, 150 mM NaCl) containing 0.05% Tween-20 (TTBS) and 5% milk (Thrive Life, UT), and then incubated with TTBS solution containing Anti-SUMO 2/3-HRP mouse monoclonal antibody (ASM23-HRP), rabbit TFII-I (abcam), rabbit Ubc9 (abcam), mouse RhoGDI (Millipore), or rabbit RhoGDI (santa cruz). Western blot membranes were incubated with primary antibody for 1 hour at room temperature (RT). Membranes were washed in TTBS 3x10 minutes, prior to secondary mouse or rabbit HRP labeled (Jackson ImmunoResearch Laboratories, PA) antibody for 1hr at RT. Bound antibodies were visualized with chemiluminescent reagent (Cytoskeleton, CO) according to the manufacturer’s directions.
Immunoprecipitation assay
Hela and A431 cells were lysed with ice-cold BlastR lysis buffer containing a cocktail NEM, and protease inhibitors (PIC02). DNA was removed by passing the lysate through the BlastR filter system (Cytoskeleton, CO). After dilution with BlastR dilution buffer, protein concentrations were determined with ADV02 and measured at 600nm OD. For tissue lysate preparation, mouse liver or heart was lysed at 1 mL of BlastR lysis buffer per 100mg of tissue. For these experiments 100mg of tissue was processed. After addition of 1mL of BlastR lysis buffer to 100mg of fresh tissue., the sample was placed into a homogenizer and 10-12 strokes were applied. The viscous tissue lysate was first passed through the BlastR filter to remove genomic DNA. Equal volume of BlastR dilution buffer was then added to the filtered lysate and gently mixed. Lysate was then spun in a microcentrifuge at 14k rpm for 15min at 4°C to pellet any remaining tissue debris. Additional BlastR dilution buffer was added to the recovered supernatant for a final dilution of 1:5. Protein concentrations were determined with ADV02 and measured at 600nm OD.
The appropriate amount of SUMO 2/3 affinity beads (ASM24-beads) or mIgG control beads (CIG01-beads) were added to 1 mg of lysate for 1-2 hr at 4°C on an end-over- end tumbler. After incubation, the affinity beads from each sample were pelleted, and washed 3X with BlastR wash buffer. Bound proteins were eluted using bead elution buffer (Cytoskeleton, CO) and detected by western immunoblotting.
References
- Enserink JM. Sumo and the cellular stress response. Cell Div. 10 4, doi: 10.1186/s13008-015-0010-1 (2015).
- Hickey CM, Wilson NR, Hochstrasser M. Function and regulation of SUMO proteases. Nat Rev Mol Cell Biol. 13 (12), 755-66, doi: 10.1038/nrm3478 (2012).
- Saitoh H, Hinchey J. Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. J Biol Chem. 275 (9), 6252-8, doi: (2000).
- Bruderer R, Tatham MH, Plechanovova A, Matic I, Garg AK, Hay RT. Purification and identification of endogenous polySUMO conjugates. EMBO Rep. 12 (2), 142-8, doi: 10.1038/embor.2010.206 (2011).
- Yang W, Paschen W. SUMO proteomics to decipher the SUMO-modified proteome regulated by various diseases. Proteomics. 15 (5-6), 1181-91, doi: 10.1002/pmic.201400298 (2015).
- Barysch SV, Dittner C, Flotho A, Becker J, Melchior F. Identification and analysis of endogenous SUMO1 and SUMO2/3 targets in mammalian cells and tissues using monoclonal antibodies. Nat Protoc. 9 (4), 896-909, doi: 10.1038/nprot.2014.053 (2014).
- Schou J, Kelstrup CD, Hayward DG, Olsen JV, Nilsson J. Comprehensive identification of SUMO2/3 targets and their dynamics during mitosis. PLoS One. 9 (6), e100692, doi: 10.1371/journal.pone.0100692 (2014).
Related Products
Signal-Seeker™ SUMOylation 2/3 Detection Kit (30 assay) (Cat. # BK162)
IgG Control Beads (Cat. # CIG01-beads)
SUMO-2/3-HRP Antibody Mouse Monoclonal (Clone 12F3) (Cat. # ASM23-HRP)
SUMOylation 2/3 Affinity Beads (Cat. # ASM24-beads)
Paclitaxel (Taxol) (Cat. # TXD01)
Protease inhibitor cocktail (Cat. # PIC02)
Precision Red Advanced Protein Assay: 1x stock (Cat. # ADV02)