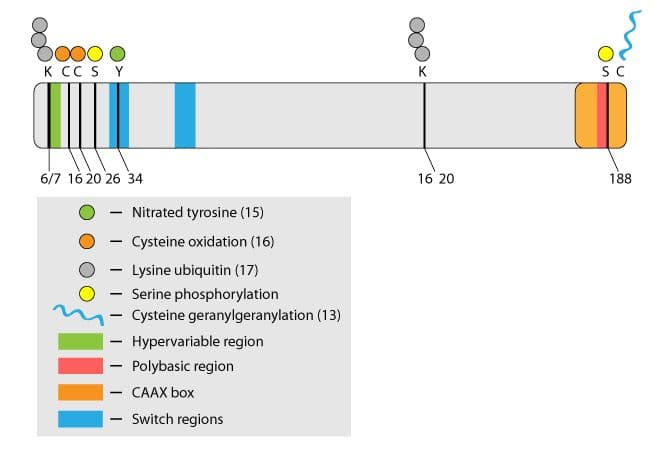
Rho family GTPases are key regulators in a wide range of physiological processes, including cell motility, cell division, and neuronal development1. Rho activity is regulated temporally and spatially by a variety of direct post-translational modifications (PTMs) that include prenylation, ubiquitination, oxidation, nitrosylation, and phosphorylation (Fig. 1). This newsletter focuses on control of RhoA function through phosphorylation. RhoA is a target for a growing number of kinases and as such, phosphorylation is emerging as a central theme in the regulation of this family of proteins2.

Mechanism of RhoA phosphorylation at Serine 188
Several kinases phosphorylate RhoA under both in vitro and in vivo conditions (Table 1). The main focus has been on PKA and PKG which phosphorylate RhoA on serine 188. There are also reports of other kinases such as SKL (Ste20-related kinase) and AMPKalpha1 (AMP-activated protein kinase subunit alpha 1) modifying RhoA (Table 1).
Early in vitro work by Lang et al.3 demonstrated that serine 188 phosphorylation by PKA deactivated RhoA. Their data supported a mechanism whereby membrane-bound RhoA-GTP is phosphorylated by PKA and the resulting membrane-bound phospho-RhoA-GTP is a substrate for Rho GDI (guanine nucleotide dissociation inhibitor) which mediates RhoA-GTP translocation to the cytosol3. This contrasts with the classical model of Rho membrane cycling in which the inactive Rho-GDP is the substrate for GDI-mediated translocation to the cytosol1. Therefore, it appears that phopshorylation provides a mechanism to inactivate Rho-GTP through disengagement from its site of action at the membrane.
| Kinase | Modified RhoA aa | Effect | Ref |
|---|---|---|---|
PKA | Ser188 | Turns off RhoA-GTP through removal from membrane to cytosol by a mechanism thought to involve Rho GDI | 3,4 |
PKA | Ser188 | Protects RhoA, particularly active RhoA from ubiquitin-mediated degradation | 7 |
PKG | Ser188 | Enhances RhoA translocation from the membrane to the cytoplasm and increases cytoplasmic RhoA levels | 6 |
PKG | Ser188 | Vasodilator action of nitric oxide | 16 |
AMPK alpha 1 | Ser188 | Vasoprotective effect of estradiol | 12 |
Mst3 | Ser26 | Reduces RhoA activity, allowing radial neuronal migration in developing brain | 17 |
The in vitro work is supported by in vivo data using transfected phosphomimetic RhoA constructs. In this system, the longstanding observation that RhoA activity is reduced in the first 15-30 minutes after plating NIH 3T3 cells onto fibronectin surfaces correlated with increased phosphorylation of RhoA at serine 1884. This correlation also agrees with reports that PKA levels are elevated during cell spreading5. Furthermore, it was shown that the PKA-mediated stress fiber collapse in 3T3 cells was completely inhibited in cells expressing the non-phosphorylatable mutant RhoA-S188A4. Similarly, stress fiber dissolution induced by constitutively active PKG in 3T3 cells was blocked in cells expressing RhoA-S188A6. These studies demonstrate that RhoA is phosphorylated in vivo by PKA and PKG. Interestingly, it has been demonstrated in vascular smooth muscle cells that RhoA serine 188 phosphorylation protects cytoplasmic phospho-RhoA-GTP from ubiquitin-mediated degradation by forming a complex with Rho GDI7. In support of this observation, stimulation of endogenous PKG activity in vascular smooth muscle leads to accumulation of RhoA-GTP in the cytoplasm7.
Thus, by directing the translocation and accumulation of active RhoA away from its site of action at the membrane, phosphorylation appears to be an alternate mechanism to the classical GTPase cycle for directly regulating RhoA. It has been suggested that a dephosphorylation event might allow the release of the cytoplasmic pool of RhoA-GTP from Rho GDI, followed by translocation back to the membrane for effector interaction and signal transduction3. A formal proof of this hypothesis has yet to be demonstrated. However, the increased affinity of phospho-Rho-GTP for Rho GDI can lead to activation of Rac1 in vascular smooth muscle cells8. This is in agreement with previously published work that demonstrates balanced regulation of Rho family proteins through sequestration of a limited Rho GDI pool9.
Physiological consequences of RhoA phosphorylation
The dramatic collapse of actin stress fibers in tissue culture cells upon induction of RhoA phosphorylation and the rescue of this phenotype by non-phosphorylatable mutants clearly demonstrates the potential of this PTM to play a significant role in the physiological regulation of RhoA. In this regard, overactivation of the RhoA/ROCK pathway is a known factor in vascular disorders such as pulmonary hypertension10. Deactivation of RhoA through serine 188 phosphorylation may represent an additional therapeutic approach to combat this group of diseases11. In this regard, estrogen, a known hypertension suppressor and vasoprotector, causes RhoA serine 188 phosphorylation through activation of AMPKalpha1 kinase12. Phosphorylation of serine 188 in RhoA has also been proposed to regulate the physiological response of cytotoxic T cells to cAMP, resulting in a block of cytolytic function. The mechanism is thought to involve altered affinity of adhesion molecules through cytoskeletal rearrangements in response to phosphorylation of RhoA by cAMP-induced PKA. This could explain the ability of cytotoxic T cells to cycle from one target to another3. In yet another example, RhoA serine 188 phosphorylation has been linked to the development of a neuroendocrine-like phenotype during androgen ablation therapy in prostate cancer. As this phenotype is associated with poor prognosis, it has been suggested that pharmacological intervention in this pathway may be a useful addition to prostate cancer therapy13.
Conclusion and future directions
There are a growing number of kinases associated with RhoA regulation. Moreover, there is a growing number of PTMs that have been demonstrated to directly regulate this protein and other members of the Rho family. As Rho proteins play such a key role in many disease states, RhoA PTMs are of great therapeutic interest14. In basic research, RhoA activity is most commonly followed by quantitating the relative increase in active GTP-bound RhoA in a cell or tissue by utilizing a variety of so-called Rho activation assays15 (http://cytoskeleton.supremeclients.com/activationassayvideo). It is becoming clear that Rho PTMs represent important additional direct regulatory mechanisms for this class of protein; further elucidation of how these mechanisms integrate and crosstalk should provide great insight into Rho family regulation in health and disease.
Related Products & Resources
NEW! Anti-Acetyl Lysine Mouse Monoclonal Antibody (Cat. # AAC01)
NEW! Anti-Ubiquitin Mouse Monoclonal Antibody (Cat. # AUB01)
RhoA G-LISA Activation Assay (Cat. # BK124)
RhoA Pull-down Activation Assay (Cat. # BK036)
Cdc42 G-LISA Activation Assay (Cat. # BK127)
Cdc42 Pull-down Activation Assay (Cat. # BK034)
Ras G-LISA Activation Assay (Cat. # BK131)
Ras Pull-down Activation Assay (Cat. # BK008)
Rac1 G-LISA Activation Assay (Cat. # BK128)
Rac1 Pull-down Activation Assay (Cat. # BK035)
Rho Inhibitor I (Cat. # CT04)
Activation Assay Video
Cytoskeleton's Small G-protein Activation Assays come in a traditional pull-down bead format or our advanced ELISA based G-LISA format. Learn which Activation Assay format is right for you by watching this video.

References
- Stankiewicz T. & Linseman D. 2014. Rho family GTPases: key players in neuronal development, neuronal survival and neurodegeneration. Front. Cell. Neurosci. doi: 10.3389/fncel.2014.00314.
- Boulter E. et al. 2012. Off the beaten paths: alternative and crosstalk regulation of Rho GTPases. FASEB J. 26, 469-479.
- Lang P. et al. 1996. Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. EMBO J. 15, 510-519.
- Ellerbrock S. et al. 2003. Serine phosphorylation negatively regulates RhoA in vivo. J. Biol. Chem. 278, 19023-19031.
- O’Connor K.L. & Mercurio A.M. 2001. Protein kinase A regulates Rac and is required for growth factor-stimulated migration of carcinoma cells. J. Biol. Chem. 276, 47895-47900.
- Sawada N. et al. 2001. cGMP-dependent protein kinase phosphorylates and inactivates RhoA. Biochem. Biophys. Res. Commun. 28, 798-805.
- Rolli-Derkinderen M. et al. 2005. Phosphorylation of serine 188 protects RhoA from ubiquitin/proteasome-mediated degradation in vascular smooth muscle cells. Circ. Res. 96, 1152-1160.
- Rolli-Derkinderen M. et al. 2010. RhoA phosphorylation induces Rac1 release from guanine dissociation inhibitor alpha and stimulation of vascular smooth muscle cell migration. Mol. Cell Biol. 30, 4786-4796.
- Boulter E. et al. 2010. Regulation of Rho GTPase crosstalk, degradation and activity by RhoGDI 1. Nature Cell Biol. 12, 477-483.
- Montani D. et al. 2014. Targeted therapies in pulmonary arterial hypertension. Pharmocol. Ther. 141, 172-191.
- Zhao Y. et al. 2012. Protein kinase G-1 deficiency induces pulmonary hypertension through RhoA/Rho kinase activation. Am. J. Path. 180, 2268-2275.
- Gayard M. et al. 2011. AMPK alpha 1-induced RhoA phosphorylation mediates vasoprotective effect of estradiol. Artherioscler. Thromb. Vasc. Biol. 31, 2634-2642.
- Jones S. & Palmer T. 2012. Protein kinase A-mediated phosphorylation of RhoA on serine 188 triggers the rapid induction of a neuroendocrine-like phenotype in prostate cancer epithelial cells. Cell. Signal. 24, 1504-1514.
- Konstantinopoulos P. et al. 2007. Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat. Rev. 6, 541-555.
- http://cytoskeleton.supremeclients.com/activationassayvideo.
- Sauzeau V. et al. 2000. Cyclic GMP-dependent protein kinase signaling pathway inhibits RhoA-nduced calcium sensitization of contraction in vascular smooth muscle. J. Biol. Chem. 275, 21722-21729.
- Tang J. et al. 2014. Cdk5-dependent Mst3 phosphorylation and activity regulate neuronal migration through RhoA inhibition. J. Neurosci. 34, 7425-7436.
- Rafikov R. et al. 2014. Lipopolysaccharide-induced lung injury involves the nitration-mediated activation of RhoA. J. Biol. Chem. 289, 4710-4722.
- Heo J. & Campbell S.L. 2005. Mechanism of redox-mediated guanine nucleotide exchange on redox-active Rho GTPases. J. Biol. Chem. 280, 31003-31010.
- Sahai E. et al. 2007. Smurf1 regulates tumor cell plasticity and motility through degradation of RhoA leading to the localized inhibition of contractility. J. Cell Biol. 176, 35-42.