YAP1 grabs the spotlight in oncogenic Ras addiction
YAP1 and the Hippo Pathway
The Hippo signal transduction pathway plays a critical role in the regulation of organ size through the coordinated modulation of cell fate1. The core pathway members include two sets of serine/threonine kinases (MST1/2 and LATS1/2) that act in succession to shut down pro-growth signaling through the phosphorylation and “inactivation” of the transcriptional co-activators YAP and TAZ2,3 (Fig. 1). YAP and TAZ are paralogous proteins that have overlapping, but not identical, functions in regulating cell growth, differentiation, and apoptosis4. For simplicity, this newsletter will focus specifically on YAP1; however most of what will be discussed has been shown to be true for TAZ. The MST1/2 — LATS1/2 — YAP1 signaling axis is activated as a result of a cell's response to physical changes in the extracellular environment, including cell adhesion, cell-cell contacts, and extracellular matrix stiffness, etc.5. Notably, several signaling pathways intersect with the Hippo pathway to ultimately control YAP1 activity6. As a result of Hippo pathway activation, YAP1 is phosphorylated by LATS1/2 on multiple serine residues (S61, S109, S127, S164, S381), which subsequently leads to the binding of 14-3-3 to phosphorylated S127 and the cytoplasmic retention of YAP13. Additionally, the phosphorylation of S381 facilitates the binding of CK1δ/ε, leading to further serine phosphorylations (S384, S387) that promote the interaction of YAP1 with the SCFbTRCP ubiquitin ligase complex. This chain of events results in YAP1 ubiquitination and subsequent proteosome-dependent degradation7. In the absence of Hippo pathway signaling, YAP1 is shuttled into the nucleus where its primary nuclear partners are the TEAD transcription factors4. Importantly, there are several other transcription factors that have been shown to partner with YAP1 in a context-dependent manner (e.g., SMADs, ß-catenin, Fos, E2F, etc.)4,8,9 (Fig. 1).

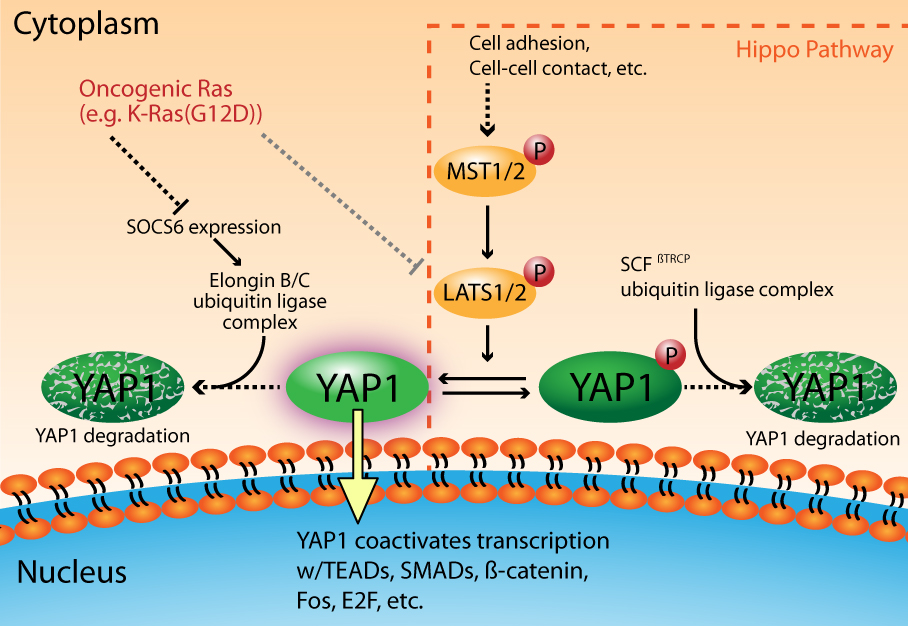
Figure 1. YAP1 activation is a critical component of oncogenic Ras Signaling
The role of YAP1 in cancer is complex
The overexpression of YAP1 in non-transformed cells leads to its nuclear accumulation, resulting in adhesion-independent cell growth, epithelial-to-mesenchymal transition (EMT), suppression of apoptosis, and growth factor-independent proliferation, all hallmarks of neoplastic cells2,10. While a direct cause and effect relationship has not been firmly established for YAP1 and the development of cancer, it is clear that in most contexts elevated YAP1 activity would support neoplastic behavior. Consistent with the in vitro YAP1 overexpression studies, several cancers have been shown to exhibit elevated YAP1 expression2,11. Moreover, elevated YAP1 activity has been correlated to a poor prognosis in several cancers12,13. The etiology of YAP1 activation in cancer is varied and often results from the dysregulation of the Hippo pathway and/or the amplification of the 11q22 genomic locus that encompasses the YAP1 gene10,14,15. In contrast to its association with cancer progression, there are cases where YAP1 appears to act as a tumor suppressor (e.g., breast cancer and some colon cancers)16,17. One clue to this apparent paradox for YAP1 activity comes from studies looking at the phosphorylation of YAP1 on two tyrosine residues (Y357 and Y391) by kinases in well-established pro-oncoprotein signaling pathways. The tyrosine kinase YES1 phosphorylates YAP1 on Y357, thereby fostering its interaction with ß-catenin to drive Wnt pathway signaling18, a known oncogenic signaling pathway19. Conversely, the tyrosine kinase c-Abl phosphorylates YAP1 on Y391 in response to DNA damage, leading to its association with p73 and the recruitment of p300 to stimulate the transcription of proapoptotic genes20 — a decidedly tumor suppressive activity. In both cases, YAP1 is active and nuclear, but the diametrically opposed transcription programs that are activated are dictated by signal dependent interactions between YAP1 and different nuclear binding partners.
YAP can overcome oncogenic Ras addiction
Recent evidence has shed light on the importance of YAP1 in Ras-dependent cancers8,9,21,22. Gain of function mutations in Ras (H-, K-, or N-Ras) have been observed in 33% of all human cancers23,24. These mutant Ras isoforms are dominant drivers of oncogenesis and the resultant cancers are highly dependent, or “addicted”, to mutant Ras signaling for their survival25,26. YAP1 has been identified as a critical factor in oncogenic Ras signaling that allows cells to overcome their Ras addiction8,9,22. Ras signaling modulates YAP1 activity at multiple levels, including the inactivation of LATS kinases27, and through the regulation of YAP1 turnover by a ubiquitin ligase complex that is distinct from the SCFßTRCP complex employed by the Hippo signaling pathway22 (Fig. 1). In the latter case, Ras signaling downregulates the expression of SOCS6, which is the YAP1 substrate recognition component of the Elongin B/C ubiquitin ligase complex22. This ultimately leads to increased levels of YAP1, its nuclear accumulation, and the activation of pro-oncogenic transcription that is mediated through its binding partners Fos and TEAD2/E2F8,9,22 (Fig. 1).
Conclusion
The finding that YAP1 can overcome oncogenic Ras addiction, and that its activity is elevated in several cancers, implies that therapeutics targeting Ras alone will likely be met with resistance that is mediated in part by YAP1. Moreover, YAP1 can act in a cell context dependent manner in either a pro-oncogenic or tumor suppressive capacity, suggesting that there may be therapeutic opportunities to promote its tumor suppressive behavior when developing drugs to target oncogenic Ras. Importantly, the activity of YAP1 is significantly modulated by several post-translational modifications (PTMs) (i.e., serine and tyrosine phosphorylation and ubiquitination). At Cytoskeleton, we have several products to further research in this area, including Ras activation assays, PTM antibodies, and GEF assays.
Related Products & Resources
NEW! Anti-Acetyl Lysine Mouse Monoclonal Antibody (Cat. # AAC01)
NEW! Anti-Phosphotyrosine Mouse Monoclonal Antibody (Cat. # APY03)
NEW! Anti-Ubiquitin Mouse Monoclonal Antibody (Cat. # AUB01)
Ras G-LISA Activation Assay (Cat. # BK131)
Ras Pull-down Activation Assay (Cat. # BK008)
RhoGEF Exchange Assay Biochem Kit (in vitro fluorescence format) (Cat. # BK100)
SOS exchange domain (564-1049) protein (Cat. # CS-SOS1)
Activation Assay Video
Cytoskeleton's Small G-protein Activation Assays come in a traditional pull-down bead format or our advanced ELISA based G-LISA format. Learn which Activation Assay format is right for you by watching this video.

References
- B. Zhao et al. 2015. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat. Cell Biol. 13, 877-883.
- J. Dong et al. 2007. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 130, 1120-1133.B.
- Zhao et al. 2007. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 21, 2747-2761.
- K. Wang et al. 2009. YAP, TAZ, and Yorkie: a conserved family of signal-responsive transcriptional coregulators in animal development and human disease. Biochem. Cell Biol. 87, 77-91.
- B.C. Low et al. 2014. YAP/TAZ as mechanosensors and mechanotransducers in regulating organ size and tumor growth. FEBS Lett. 588, 2663-2670.
- K.D. Irvine. 2012. Integration of intercellular signaling through the Hippo pathway. Semin. Cell Dev. Biol. 23, 812-817.
- B. Zhao et al. 2010. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCFb-TRCP. Genes Dev. 24, 72-85.
- D.D. Shao et al. 2014. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell. 158, 171-184.
- A. Kapoor et al. 2014. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell. 158, 185-197.
- M. Overholtzer et al. 2006. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc. Natl. Acad. Sci. U.S.A. 103, 12405-12410.
- D.M. Lam-Himlin et al. 2006. The hippo pathway in human upper gastrointestinal dysplasia and carcinoma: a novel oncogenic pathway. Int. J. Gastrointest. Cancer. 37, 103-109.
- M.Z. Xu et al. 2009. Yes-associated protein is an independent prognostic marker in hepatocellular carcinoma. Cancer. 115, 4576-4585.
- C.A. Hall et al. 2010. Hippo pathway effector Yap is an ovarian oncogene. Cancer Res. 70, 8517-58525.
- L. Zender et al. 2006. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell. 125, 1253-1267.
- T. Muramatsu et al. 2011. YAP is a candidate oncogene for esophageal squamous cell carcinoma. Carcinogenesis. 32, 389-398.
- M. Yuan et al. 2008. Yes-associated protein (YAP) functions as a tumor suppressor in breast. Cell Death Differ. 15, 1752-1759.
- E.R. Barry et al. 2013. Restriction of intestinal stem cell expansion and the regeneration response by YAP. Nature. 493, 106-110.
- J. Rosenbluh et al. 2012. b-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell. 151, 1457-1473.
- J. Rosenbluh et al. 2014. Genomic insights into WNT/b-catenin signaling. Trends Pharmacol. Sci. 35, 103-109.
- D. Levy et al. 2008. Yap1 phosphorylation by c-Abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Mol. Cell. 29, 350-361.
- W. Zhang et al. 2014. Downstream of mutant KRAS, the transcriptional regulator YAP is essential for neoplastic progression to pancreatic ductal adenocarcinoma. Sci. Signal. 7, ra42.
- X. Hong et al. 2014. Opposing activities of the Ras and Hippo pathways converge on regulation of YAP protein turnover. EMBO J. 33, 2447-2457.
- A.T. Baines et al. 2011. Inhibition of Ras for cancer treatment: the search continues. Future Med Chem. 3, 1787-1808.
- A.E. Karnoub and R.A. Weinberg. 2008. Ras oncogenes: split personalities. Nat. Rev. Mol. Cell Biol. 9, 517-531.
- I.B. Weinstein. 2002. Cancer. Addiction to oncogenes – the Achilles heal of cancer. Science. 297, 63-64.
- F. McCormick. 2011. Cancer therapy based on oncogene addiction. J. Surg. Oncol. 103, 464-467.
- B.V. Reddy and K.D. Irvine. 2013. Regulation of Hippo signaling by EGFR-MAPK signaling through Ajuba family proteins. Dev. Cell. 24, 459-471.