Neurodegeneration: Rhes, SUMOylation, and Huntington's Disease
The small G-protein Rhes (Ras Homolog Enriched in Striatum) is a 266 amino acid protein found predominantly in striatum, and to a lesser extent, the cerebral cortex (Falk et al., 1999). Recent research has revealed that this GTPase may be key to understanding the paradoxical finding that while many different types of cells throughout the brain and body express wild-type and mutant huntingtin protein (mHTT), striatal neurons (and to a degree, cortical neurons) have a selective vulnerability in Huntington’s disease (HD) (Harrison, 2012; Harrison and LaHoste, 2013). HD is an autosomal dominant neurodegenerative disorder characterized by >35 CAG repeats in the huntingtin gene, resulting in expression of mHTT that contains multiple glutamine residues (a.k.a. polyglutamine or polyQ repeats).
HD is incurable and untreatable, causing deficits in movement, cognition, and mood. Neurons expressing mHTT are selectively lost in striatal and cortical neurons, despite the ubiquitous expression of the mutant protein in cells throughout the brain and peripheral tissues (Harrison, 2012; Harrison and LaHoste, 2013). mHTT exists in two forms in neurons: 1. toxic soluble monomer form and 2. aggregates or inclusions which sequester the protein. These insoluble aggregates are believed to be neuroprotective (Saudou et al., 1998; Gong et al., 2008; Lu and Palacino, 2013) (Fig. 1). mHTT is modified by multiple post-translational modifications (PTMs), including SUMOylation. This PTM causes deaggregation of the inclusions, releasing the soluble mHTT monomers and increasing neurotoxicity (Steffan et al., 2004) (Fig. 1).

Importantly, expression of mHTT by itself does not confer HD pathology (Zeron et al., 2002; Okamoto et al., 2009). Thus, there are other proteins or signaling pathways involved in mediating mHTT-induced neurodegeneration. A major breakthrough in HD research came with the discovery that Rhes preferentially binds mutant over wild-type HTT and acts as a SUMO E3 ligase to SUMOylate mHTT in vitro (Subramaniam et al., 2009) (Fig. 1). SUMOylation of mHTT by Rhes increases levels of soluble mHTT and augments mHTT-mediated neurotoxicity via deaggregation of inclusions and/or reduced inclusion formation (Subramaniam et al., 2009). Rhes is also SUMOylated itself (Subramaniam et al., 2009). As would be expected, co-expression of Rhes and a mHTT protein reduced neuron viability, while silencing of the Rhes gene protected the cells from mHTT-mediated neurotoxicity in an in vitro neuron model of HD (Seredenina et al., 2011) (Fig. 1). Interestingly, mHTT is not the only protein targeted by Rhes for SUMOylation as Rhes enhances cross-SUMOylation between the SUMOylating enzymes E1 and Ubc9 and Rhes knockout mice have reduced levels of general SUMOylation in their striata (Subramaniam et al., 2010).
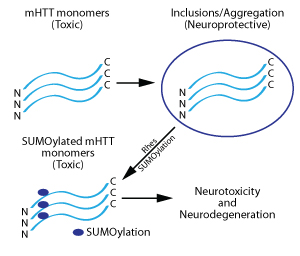
Figure 1: Processing of mHTT protein in neurons and its further modification by Rhes, leading to neurotoxicity and neurodegeneration.
The role of Rhes in HD was further confirmed in vivo with a HD transgenic mouse model crossed with Rhes knockout mice. These double transgenic mice experienced a significantly delayed and decreased onset of HD-associated symptoms (Baiamonte et al., 2013). Similarly, a striatal specific in vivo HD model was not reproduced in Rhes knockout mice (Mealer et al., 2013). This finding demonstrates that Rhes depletion protects neurons from the cell death and motor deficits induced by the HD-causing treatment in normal mice (Mealer et al., 2013).
A study examining the mechanisms mediating the selective neurodegeneration of striatal neurons has focused on the differential activation of synaptic versus extrasynaptic NMDA receptors (Okamoto et al., 2009). This study also shed light on the role of Rhes in NMDA receptor-mediated mHTT neurotoxicity. The authors found that synaptic NMDA receptor activity is necessary for, and in fact promotes, the formation of mHTT inclusions (Okamoto et al., 2009). The presence of mHTT, compared to WT HTT, increased a neuron’s vulnerability to excitotoxic insult (Zeron et al., 2002; Okamoto et al., 2009). Blockade of the extrasynaptic NMDA receptors protected mHTT-expressing neurons from excitotoxicity (Okamoto et al., 2009). This neuroprotective effect involves Rhes, as blocking the extrasynaptic NMDA receptors decreased Rhes protein levels, implicating elevated Rhes levels in mHTT-mediated neurotoxicity during stimulation of extrasynaptic NMDA receptors (Okamoto et al., 2009). These findings suggest that selective blockade of extrasynaptic NMDA receptors could serve as a beneficial therapy for protecting neurons susceptible to HD.
As additional tools for studying the PTMs of proteins, including GTPases, become available, advances in our understanding of a cell’s physiological and pathological processes will follow. Tools to study PTMs will offer clear benefits to both pre-clinical and clinical researchers in neuroscience, cardiology, and oncology.
Related Research Tools
| Kit | Cat. # | Amount | ||||||
Ras G-LISA® Activation Assay, colorimetric | BK131 | 96 assays | ||||||
Ras Activation Assay, Bead Pull-down format | 20 assays | |||||||
RhoGAP Assay Biochem Kit™ | BK105 | 80-160 assays | ||||||
RhoGEF Exchange Assay Biochem Kit™ | BK100 | 60-300 assays | ||||||
Raf-RBD beads (Binds active Ras protein) | RF02-A RF02-B | 1 x 2 mg | ||||||
| G-protein Modulator | Cat. # | Amount | ||||||
Rac/Cdc42 Activator II | CN02-A CN02-B | 5 x 10 µg | ||||||
Rho Inhibitor I | CT04-A CT04-B | 1 x 20 µg | ||||||
References
- Falk J.D., Vargiu P., Foye P.E., Usui H., Perez J. et al. 1999. Rhes: A striatal-specific Ras homolog related to dexras1. J. Neurosci. Res. 57, 782-788.
- Harrison L.M. 2012. Rhes: A GTP-binding protein integral to striatal physiology and pathology. Cell Mol. Neurobiol. 32, 907-918.
- Harrison L.M. and LaHoste G.J. 2013. The role of Rhes, Ras homolog enriched in striatum, in neurodegenerative processes. Exp. Cell Res. 319, 2310-2315.
- Saudou F., Finkbeiner S., Devys D., and Greenberg M.E. 1998. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 95, 55-66.
- Gong B., Lim M.C., Wanderer J., Wyttenbach A., and Morton A.J. 2008. Time-lapse analysis of aggregate formation in an inducible PC12 cell model of Huntington’s disease reveals time-dependent aggregate formation that transiently delays cell death. Brain Res. Bull. 75, 146-157.
- Lu B. and Palacino J. 2013. A novel human embryonic stem cell-derived Huntington’s disease neuronal model exhibits mutant huntingtin (mHTT) aggregates and soluble mHTT-dependent neurodegeneration. FASEB J. 27, 1820-1829.
- Steffan J.S., Agrawal N., Pallos J., Rockabrand E., Trotman L.C. et al. 2004. SUMO modification of huntingtin and Huntington’s disease pathology. Science. 304, 100-104.
- Zeron M.M., Hansson O., Chen N., Wellington C.L., Leavitt B.R., et al. 2002. Increased sensitivity to N-Methyl-D-Aspartate receptor-mediated excitotoxicity in a mouse model of Huntington’s disease. Neuron. 33, 849-860.
- Okamoto S., Pouladi M.A., Talantova M., Yao D., Xia P. et al. 2009. Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat. Med. 15, 1407-1413.
- Subramaniam S., Sixt K.M., Barrow R., and Snyder S.H. 2009. Rhes, a striatal specific protein, mediates mutant huntingtin cytotoxicity. Science. 324, 1327-1330.
- Seredenina T., Gokce O., and Luthi-Carter R. 2011. Decreased striatal RGS2 expression is neuroprotective in Huntington’s disease (HD) and exemplifies a compensatory aspect of HD-induced gene regulation. PLoS ONE. 6:e22231.
- Subramaniam S., Mealer R.G., Sixt K.M., Barrow R.K., Usiello A., et al. 2010. Rhes, a physiologic regulator of sumoylation, enhances cross-sumoylation between the basic sumoylation enzymes E1 and Ubc9. J. Biol. Chem. 285, 20428-20432.
- Baiamonte B.A., Lee F.A., Brewer S.T., Spano D., and LaHoste G.J. 2013. Attenuation of Rhes activity significantly delays the appearance of behavioral symptoms in a mouse model of Huntington’s disease. PLoS ONE. 8:e53606.
- Mealer R.G., Subramaniam S., and Snyder S.H. 2013. Rhes deletion is neuroprotective in the 3-nitropropionic acid model of Huntington’s disease. J. Neurosci. 33, 4206-4210.