Paclitaxel induces post-translational modifications of RhoGDI alpha: a potential mechanism to regulate RhoA activity
Henrick Horita, Andy Law, and Kim Middleton
American Society for Cell Biology, 2017, Poster B624
Cytoskeleton Inc., Denver, CO 80223
Abstract
There is evidence in the literature of crosstalk between the microtubule and microfilament systems including studies showing that depolymerization of microtubules regulate stress fiber formation by RhoA dependent mechanisms. A recent study showed that low dose of paclitaxel inhibited RhoA function; however, the mechanism by which microtubule stabilization or destabilization regulates RhoA activity is not fully understood. RhoA activity is regulated by several mechanisms including RhoGDIα interaction, which inhibits RhoA activity through binding and spatial localization. RhoGDIα in-turn is highly regulated; for example, it can be modified by post-translational modifications (PTMs) such as SUMOylation and acetylation. Specifically, acetylation of RhoGDIα has been shown to prevent binding to RhoA thus promoting RhoA activity; conversely, SUMO-1 modification of RhoGDIα enhances RhoGDIα -RhoA complex formation and inhibits RhoA activity.
Paclitaxel has been shown to alter the SUMO 2/3 state of RhoGDIα in mitosis using overexpression systems. Thus, we asked the question of whether paclitaxel regulates RhoGDIα PTMs as a mechanism to alter RhoA activity. Physiologic and endogenous SUMO 2/3 modified RhoGDI was detected in response to 6hr paclitaxel treatment in Hela and A431 cells. The RhoGDIα SUMO 2/3 modification was validated with removal of desumoylase inhibitor, as well as inhibition of the SUMO ligase with 2-D08. A timecourse of paclitaxel treatment was performed followed by measurements of both SUMO 2/3 modifciation of RhoGDIα and RhoA activity to better correlate SUMO 2/3 changes with a change in downstream RhoA activity. Surprisingly, there was a rapid increase in RhoA activity at early time points prior to a decrease observed at 6 hrs of paclitaxel treatment. The significant decrease in RhoA activity observed at 6hr correlated with the increase in SUMO 2/3 RhoGDIα modification, and suggests that SUMO 2/3 modification of RhoGDIα may have a similar role to SUMO-1 modification of RhoGDIα. As RhoGDIα SUMO 2/3 levels were not detectable at early time points, this modification could not be responsible for the early change in RhoA activity. However, a paclitaxel induced increase in acetylation of RhoGDIα did correlate with the increased RhoA activity.


Of particular interest, the acetylated RhoGDIα signal disappeared as the SUMO 2/3 signal appeared in A431 cells, which supports previous in vitro findings of competition between these two PTMs on RhoGDIα. Importantly, data shown here suggests that crosstalk between RhoGDIα acetylation and SUMO 2/3 modification occur physiologically in response to paclitaxel, and forced maintenance of acetylated RhoGDIα with TSA treatment prevented SUMO 2/3 modification of RhoGDIα in response to paclitaxel in A431 cells. Furthermore, TSA treatment can reverse paclitaxel induced RhoA activity, as well, as early morphological changes. Collectively, the data highlight the importance of PTMs and their crosstalk in regulating RhoGDIα function, and illuminate how microtubule stabilization may induce RhoGDIα -RhoA regulatory mechanisms of communication between the microtubule and microfilament cytoskeletal systems.
Results
Figure 1
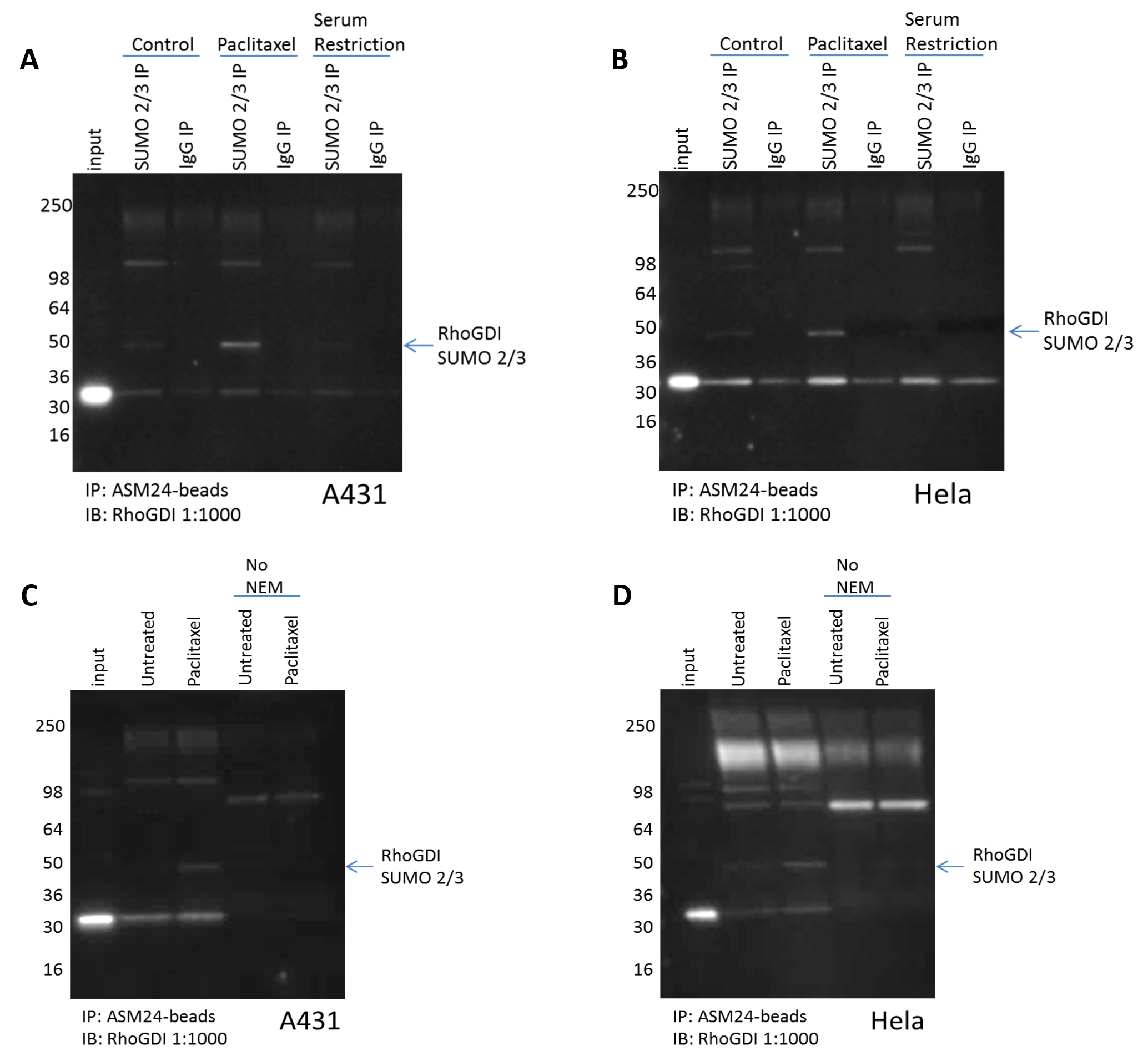
Figure 1. RhoGDI is SUMO 2/3 modified in response to Paclitaxel. Untreated, paclitaxel 100nM, or serum restricted (0% FBS) treated (A) A431, or (B) Hela cells were lysed with BlastR lysis buffer supplemented with deSUMOylase inhibitor (NEM). 1mg of each lysate was incubated with the recommended amount of SUMO 2/3 affinity beads (Cat. # ASM24-beads) or IgG control beads (Cat. # CIG01-beads). Immunoprecipitated samples were separated by SDS-PAGE and transferred to PVDF. Western blot was performed with RhoGDI. Untreated or paclitaxel 100nM treated (C) A431, or (D) Hela cells were lysed with BlastR lysis buffer supplemented with or without deSUMOylase inhibitor (NEM). 1mg of each lysate was incubated with the recommended amount of SUMO 2/3 affinity beads (Cat. # ASM24-beads). Immunoprecipitated samples were separated by SDS-PAGE and transferred to PVDF. Western blot was performed with RhoGDI.
Figure 2
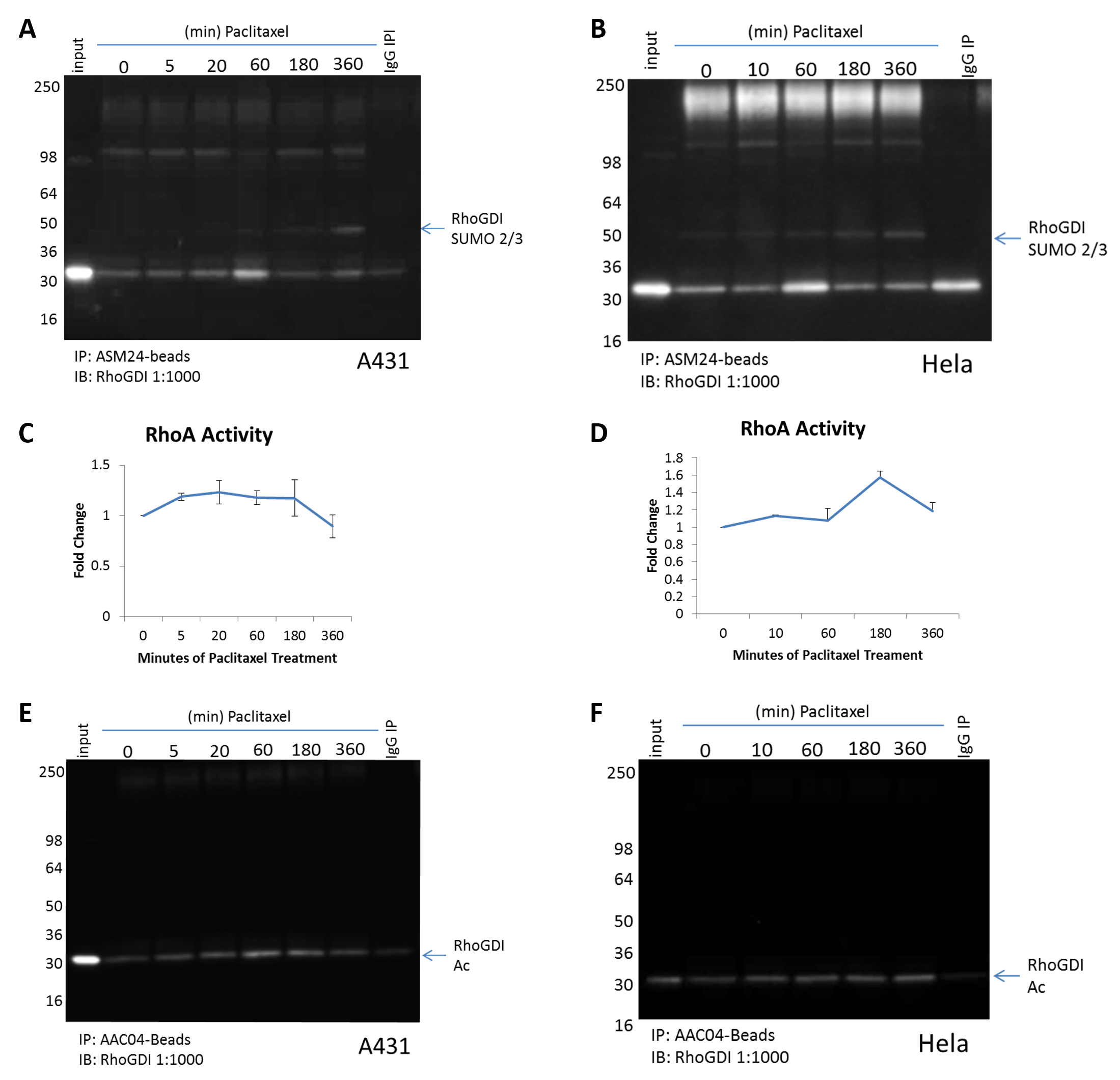
Figure 2. Paclitaxel induced RhoA activity correlates with changes in RhoGDI PTMs. (A) A431 cells or (B) Hela cells were treated over a timecourse with paclitaxel 100nM. Cells were lysed with BlastR lysis buffer supplemented with deSUMOylase inhibitor (NEM) and TSA. 1mg of each lysate was incubated with SUMO 2/3 affinity beads (Cat. # ASM24-beads) or IgG control beads (Cat. # CIG01-beads). Immunoprecipitated samples were separated by SDS-PAGE and transferred to PVDF. Western blot was performed with RhoGDI. (C) A431 cells or (D) Hela cells were treated over a timecourse with paclitaxel 100nM. RhoA G-LISA activation assays were used to measure RhoA activity. (E and F) 1mg of each lysate from A and B were incubated with acetyl-lysine affinity beads (Cat. # AAC04-beads) or control beads (Cat. #CIG02-beads). Immunoprecipitated samples were separated by SDS-PAGE and transferred to PVDF. Western blot was performed with RhoGDI.
Figure 3

Figure 3. Crosstalk between RhoGDI acetylation and SUMO 2/3 modification. (A) A431 cells or (B) Hela cells were pre-treated with TSA 1mM for 30 minutes followed by either DMSO or paclitaxel 100nM for 6 hours.. Cell lysates were prepare as in figure 2. 1mg of each lysate was incubated with SUMO 2/3 affinity beads (Cat. # ASM24-beads) or IgG control beads (Cat. # CIG01-beads). Immunoprecipitated samples were separated by SDS-PAGE and transferred to PVDF. Western blot was performed with RhoGDI. (C and D) 1mg of each lysate from A and B were incubated with acetyl-lysine affinity beads (Cat. # AAC04-beads) or control beads (Cat. #CIG02-beads). Immunoprecipitated samples were separated by SDS-PAGE and transferred to PVDF. Western blot was performed with RhoGDI.
Figure 4
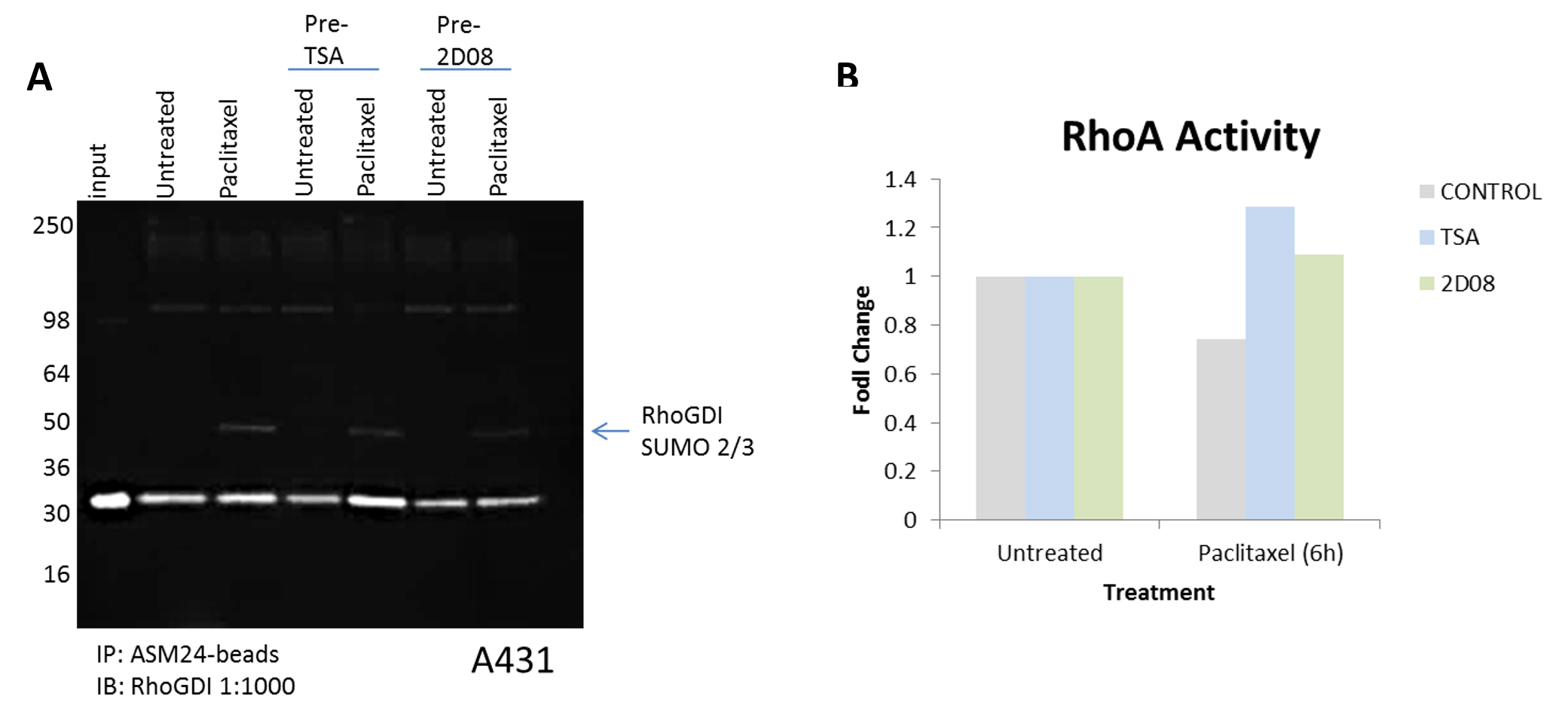
Figure 4. Regulating RhoGDI‘s PTM state alters RhoA activity. (A) A431 cells were pre-treated with TSA or 2-D08 for 30 minutes followed by either DMSO or paclitaxel 100nM for 6 hours. Cell lysates were prepare as in figure 2. 1mg of each lysate was incubated with SUMO 2/3 affinity beads (Cat. # ASM24-beads) or IgG control beads (Cat. # CIG01-beads). Immunoprecipitated samples were separated by SDS-PAGE and transferred to PVDF. Western blot was performed with RhoGDI. (B) A431 cells were pre-treated with TSA or 2-D08 for 30 minutes followed by either DMSO or paclitaxel 100nM for 6 hours. RhoA G-LISA activation assays were used to measure RhoA activity.
Figure 5

Figure 5. Paclitaxel induced cell morphological changes are suppressed with TSA treatment. (A) A431 cells were pre-treated with TSA 1mM for 30 minutes followed by either DMSO or paclitaxel 100nM for 6 hours. Representative images were taken of actin fibers that were visualized using Acti-Stain 488 (phalloidin) (Cat. # PHDG1-A). (B) A431 cells were pre-treated with TSA 1mM for 30 minutes followed by either DMSO or paclitaxel 100nM for 6 hours. Representative bright field images were taken at 4x and 10x magnification.
Conclusions
-
RhoGDIα is endogenously modified by SUMO 2/3 in a response to paclitaxel.
-
RhoGDIα PTM modifications, SUMOylation and acetylation, alters RhoA activity
-
SUMO 2/3 and acetylation PTM crosstalk exists on RhoGDIα; however, the significance of this crosstalk appears to be cell type dependent
-
Altering RhoGDIαand/or RhoA activity through PTM inhibitors alters paclitaxel induced morphological changes. However, more specific PTM inhibitors are necessary to define which modified proteins are most essential
-
Additional PTM research tools are necessary; for example, site specific acetylation and SUMOylation antibodies. These tools could provide insight into the cell specific crosstalk

Related Products
Signal-Seeker™ Phosphotyrosine Detection Kit (Cat. # BK160)
Signal-Seeker™ Ubiquitination Detection Kit (Cat. # BK161)
Signal-Seeker™ SUMOylation 2/3 Detection Kit (Cat. # BK162)
Signal-Seeker™ Acetyl-Lysine Detection Kit (Cat. # BK163)
BlastR Rapid Lysate Prep Kit (Cat. # BLR01)
Paclitaxel (Taxol) (Cat. # TXD01)
Acti-stain™ 488 phalloidin (Cat. # PHDG1)
RhoA G-LISA Activation Assay Kit (Colorimetric format) 96 assays (Cat. # BK124)