June Newsletter: Actin Methionine Oxidation: The Next Level of Dynamic Regulation
Actin PTM Background
Actin is a well-characterized, abundant, and essential cytoskeletal protein. Its dynamic properties allow it to shift between monomeric (G-actin) and polymeric (F-actin) states, which is vital for many cellular processes. Actin’s dynamicity and function is regulated by many internal and external cues that are facilitated by actin binding proteins (ABPs), signal transducers, and others. Additionally, several studies now indicate that actin itself is highly modified by post-translational modifications (PTMs); furthermore, intensive studies of specific actin PTMs have detailed their effect on actin dynamics, ABP interactions, and actin-dependent physiology1,2. For example, actin N-terminal acetylation, lysine acetylation, arginylation, SUMOylation, and ubiquitination have been studied. Here, current studies on physiologic oxidation of actin at methionine (Met)44 and Met47 are summarized.
Actin Oxidation: Focus on Methionine Sulfoxide (MetO)

The oxidation field in general has focused on pathological oxidation (oxidative stress) induced by naturally formed oxidants like H2O2, and while it was initially hypothesized that oxidation was “toxic”, it is now known that oxidation is a signaling mechanism for both pathological and physiological events3. Most of this work related to reactive oxygen species (ROS) focused on thiol-based cysteine (Cys) modifications. This is also true in the case of actin, whereby, most of the early work on actin oxidation utilized H2O2 treatment and resulted in altered F-actin content and polymerization activity as defined by enhanced lag time, slower polymerization rate, and lower polymerization extent4-6. Further investigation showed that H2O2-induced oxidation of actin initially targeted Cys374, but also targeted several Mets, including, Met44, Met47, Met176, Met190, Met269, and Met355 in vitro7. MetO oxidation happens in vivo under normal or stress conditions; however, Manta and Gladsyshev suggested that ROS-induced MetO formation in vivo occurs very inefficiently relative to the kinetics of reducing enzymes like MsrA8. Thus, the question of whether actin’s methionines can become oxidized enzymatically in vivo still remained unanswered. A seminal study by the Terman group identified a role for the enzyme, MICAL (molecule interacting with CasL), in mediating oxidation of Met44 and Met47 of actin in vitro9. These in vitro studies showed that MetO at Met44 was sufficient to promote both severing of filaments and decreased polymerization. Additionally, mis-regulation of actin MetO produced profound morphological consequences as overexpression of MICAL in Drosophila resulted in deformed bristle formation but was rescued with M44L actin mutants; thereby, defining its role in vivo9,10. A recent study identified a physiologic role for MICAL-1 oxidation of F-actin by determining that this mechanism is critical for depolymerizing actin during the terminal steps of cytokinesis11. These studies shed light on the physiologic importance that actin MetO has on actin’s molecular, cellular, and morphologic functions.
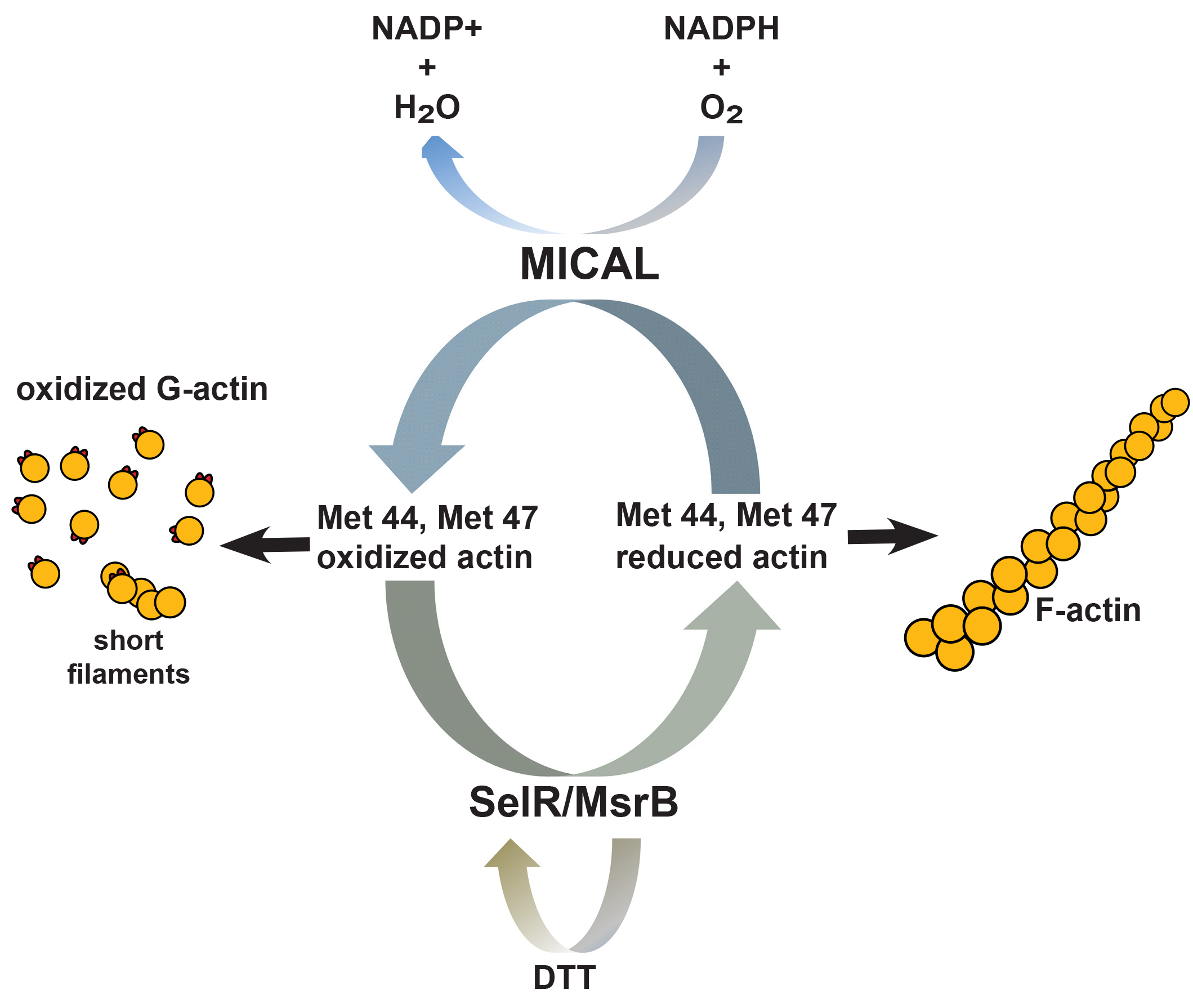
Regulatory Mechanisms of Actin Oxidation
MICAL is an intracellular flavoprotein monoxygenase, conserved from insects to mammals, that functions as a catalyst for oxidation-reduction (redox) reactions12. There are three MICAL family members in mammals, and all of them can oxidize actin but with different kinetics and spatial localization and regulation13. Terman’s group showed that MICAL interacts with actin and uses NADPH as a cofactor to oxidize actin at Met44 and Met479. The Met44 residue resides in actin’s D-loop of subdomain 2 which is critical for actin subunit contacts14. When oxidized, Met44 becomes negatively charged which interferes with actin monomer-monomer interactions; thus, promoting F-actin severing and depolymerization. Importantly, MICAL-mediated effects on actin do not occur through a diffusible oxidant like H2O2, as reductants like DTT did not alter MICAL activity, and MICAL needed to be in close proximity to actin9.
Recent studies discovered that MICAL-mediated MetO of actin is reversed by the SelR/MsrB family of methionine sulfoxide reductase enzymes. Two groups independently identified SelR (MsrB) as the enzyme responsible for reduction of Met44 and Met47 and restoration of normal actin dynamics15,16. These studies determined that SelR/MsrB selectively reduced MICAL-oxidized actin, while another methionine sulfoxide reductase member, MsrA, did not. Since SelR/MsrB specifically reduces R-isomer MetO and MsrA specifically reduces S-isomer MetO, the groups concluded that MICAL stereo-specifically oxidized actin with MetO R-isomer. Collectively, these data describe a reversible, specific redox system that controls actin dynamics and cytoskeletal organization through regulation of a specific MetO of actin.
Actin Oxidation Physiology
Since MICALs appear to be expressed ubiquitously and the Met44 and Met47 residues of actin are highly conserved, it is likely that this mechanism of redox regulation may play a prominent role in modulating actin function in all tissue and cell types. Several studies highlight that MICAL proteins play a role both physiologically and pathologically in an array of tissues and organisms; however, whether or not these effects are facilitated through actin-dependent regulation have not been thoroughly investigated17. Following are a few examples where MICAL had a profound effect linked to its specific regulation of actin. One such study identified MICAL-2 as a regulator of nuclear G-actin levels, subsequent MRTF-A/SRF transcriptional regulation, and physiological regulation of heart development in zebrafish18. Another study investigated the effects that growth factors and chemo-repellents have on MICAL regulation of actin, and found paradoxically that effects of the chemo-repellent were amplified by growth factor signaling which had profound effects on physiological axon guidance regulation, as well as pathological tumor progression and response to treatment19. Finally, MICAL-1 is important for the development of hippocampal mossy fiber connections through F-actin regulation, and this physiological process may be important in neural disorders20. These studies and others highlight the profound effect that this small, physiologic MetO of actin can have on several distinct cellular processes.
Conclusions and Future Directions
These studies have undoubtedly laid significant groundwork towards advancing the actin PTM field through identification of this novel, reversible redox regulatory mechanism. Recent studies suggest that MICAL regulation of actin may be important for many actin-dependent cellular processes, and one must wonder how pervasive this redox mechanism is for actin biology. Along this same line of thinking, it will be interesting to determine how ABP regulation of actin works together and/or in opposition with critical PTMs like actin MetO. A recent study indicates that actin oxidation and cofilin synergize to disassemble actin21, providing clear evidence ABP and actin PTM crosstalk does exist and warrants further investigation. As investigators decipher MICAL-oxidized actin’s role in disease, it will be interesting to further define the interplay of ROS vs enzymatic actin MetO. Having useful MetO actin tools to address these types of questions will undoubtedly help researchers gain a better understanding of actin biology and whether or not MetO actin plays a role in their research models. Cytoskeleton, Inc. offers a variety of MICAL-oxidized (MOX) actin tools to help researchers incorporate this novel actin regulatory mechanism into their own research as they gain a better understanding of actin biology.
References
1. Varland S. et al. 2019. Actin post-translational modifications: The Cinderella of cytoskeletal control. Trends Biochem. Sci. DOI: 10.1016/j.tibs.2018.11.010.
2. Terman J.R. and Kashina A. 2013. Post-translational modification and regulation of actin. Curr. Opin. Cell Biol. 25, 30-38.
3. Wilson C. and Gonzalez-Billault C. 2015. Regulation of cytoskeletal dynamics by redox signaling and oxidative stress: implications for neuronal development and trafficking. Front. Cell Neurosci. 9, 381.
4. Hinshaw D.B. et al. 1986. Cytoskeletal and morphologic impact of cellular oxidant injury. Am. J. Pathol. 123, 454-464.
5. DalleDonne I. et al. 1995. H2O2-treated actin: assembly and polymer interactions with cross-linking proteins. Biophys. J. 69, 2710-2719.
6. Lassing I. et al. 2007. Molecular and structural basis for redox regulation of beta-actin. J. Mol. Biol. 370, 331-348.
7. Milzani A. et al. 2000. The oxidation produced by hydrogen peroxide on Ca-ATP-G-actin. Protein Sci. 9, 1774-1782.
8. Manta B. and Gladyshev V.N. 2017. Regulated methionine oxidation by monooxygenases. Free Rad. Biol. Med. 109, 141-155.
9. Hung R.J. et al. 2011. Direct redox regulation of F-actin assembly and disassembly by Mical. Science. 334, 1710-1713.
10. Hung R.J. et al. 2010. Mical links semaphorins to F-actin disassembly. Nature. 463, 823-827.
11. Fremont S. et al. 2017. Oxidation of F-actin controls the terminal steps of cytokinesis. Nat. Commun. 8, 14528.
12. Terman J.R. et al. 2002. MICALs, a family of conserved flavoprotein oxidoreductases, function in plexin-mediated axonal repulsion. Cell. 109, 887-900.
13. Wu H. et al. 2018. The MICALs are a family of F-actin dismantling oxidoreductases conserved from Drosophila to humans. Sci. Rep. 8, 937.
14. Grintsevich E.E. et al. 2017. Catastrophic disassembly of actin filaments via Mical-mediated oxidation. Nat. Commun. 8, 2183.
15. Lee B.C. et al. 2013. MsrB1 and MICALs regulate actin assembly and macrophage function via reversible stereoselective methionine oxidation. Mol Cell. 51, 397-404.
16. Hung R.J. et al. 2013. SelR reverses Mical-mediated oxidation of actin to regulate F-actin dynamics. Nat. Cell Biol. 15, 1445-1454.
17. Wilson C. et al. 2016. Actin filaments-A target for redox regulation. Cytoskeleton (Hoboken). 73, 577-595.
18. Lundquist M.R. et al. 2014. Redox modification of nuclear actin by MICAL-2 regulates SRF signaling. Cell. 156, 563-576.
19. Yoon J. et al. 2017. Amplification of F-Actin disassembly and cellular repulsion by growth factor signaling. Dev. Cell. 42, 117-129.
20. Van Battum E.Y. et al. 2014. The intracellular redox protein MICAL-1 regulates the development of hippocampal mossy fibre connections. Nat. Commun. 5, 4317.
21. Grintsevich E.E. et al. 2016. F-actin dismantling through a redox-driven synergy between Mical and cofilin. Nat. Cell Biol. 18, 876-885.
Related Products
MOX Actin Products
Actin Products
Actin protein (>99% pure): bovine cardiac muscle (Cat. # AD99)
Actin protein (>99% pure): chicken gizzard muscle (Cat. # AS99)
Actin protein (pre-formed filaments): rabbit skeletal muscle (Cat. # AKF99)
Actin protein (>95% pure): rabbit skeletal muscle (Cat. # AKL95)
Actin protein (>99% pure): rabbit skeletal muscle (Cat. # AKL99)
Actin protein (>99% pure): human platelet (Cat. # APHL99)
Actin protein (rhodamine): human platelet (Cat. # APHR)
Actin protein (rhodamine): rabbit skeletal muscle (Cat. # AR05)
Spirochrome SiR-Actin Kit (Cat. # CY-SC001)
Spirochrome SiR700-Actin Kit (Cat. # CY-SC013)
Acti-stain 488 Phalloidin (Cat. # PHDG1)
Acti-stain 555 Phalloidin (Cat. # PHDH1)
Acti-stain 670 Phalloidin (Cat. # PHDN1)
Rhodamine Phalloidin (Cat. # PHDR1)
Actin Biochem Kits
Actin Binding Protein Spin-Down Assay Biochem Kit: rabbit skeletal muscle actin (Cat. # BK001)
Actin Binding Protein Spin-Down Assay Biochem Kit: human platelet actin (Cat. # BK013)
Actin Polymerization Biochem Kit (fluorescence format): rabbit skeletal muscle actin (Cat. # BK003)